A.4 Lab #1: .pdb Files
The first lab deals with .pdb files.
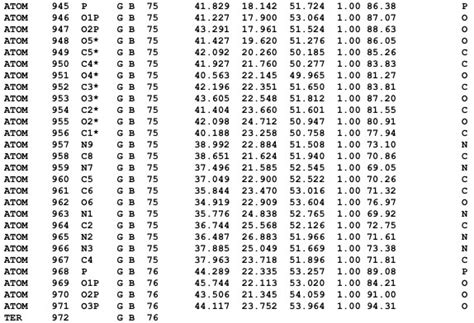
Figure A.1: Example Structure of a .pdb File
The acronym “pdb” stands for the Protein Data Bank. .pdb files are used to depict structures in the Protein Data Bank and are also read and written by many programs.
In this lab, you are given the official 1996 documentation for PDB files. You are to complete the following tasks:
A.4.1 Task #1: examining a .pdb file
In the “Full Text” section of the advanced search feature, enter a random keyword related to protein function (e.g., ribosome). For no particular reason, I chose to type “shock”:
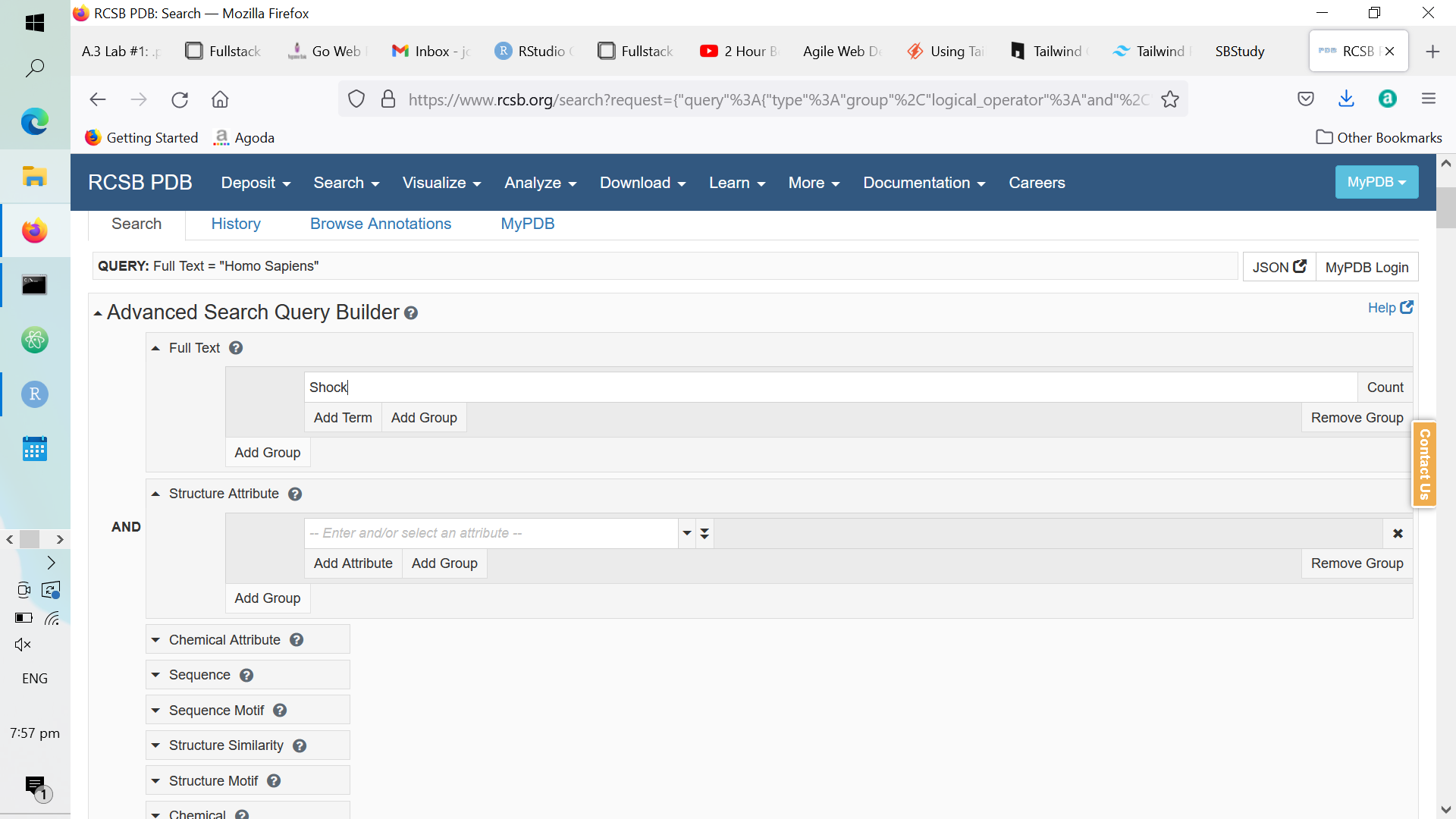
Figure A.2: Example Text in the ‘Full Text’ parameter
Thereafter, scroll down to the “Refinements” side bar. From the side bar, select the “Homo sapiens” and the “X-RAY DIFFRACTION” options like so:
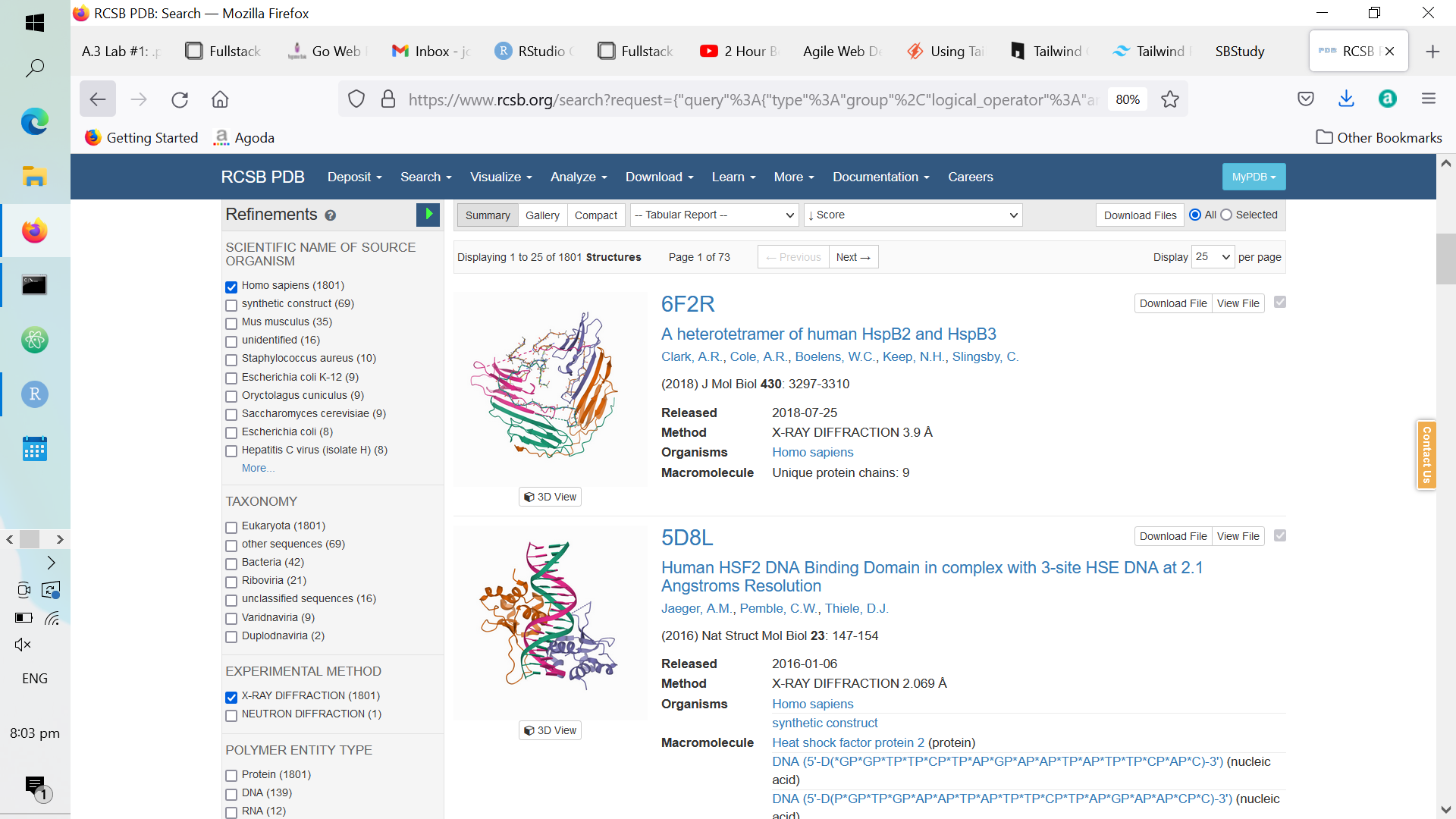
Figure A.3: Refinements to Select in the ‘Refinement’ Sidebar
Be sure to hit the play button to the top right of the refinement side bar to enhance your search queries!
Once you’ve done the above, click on any protein; thereafter, download its .pdb file like so:
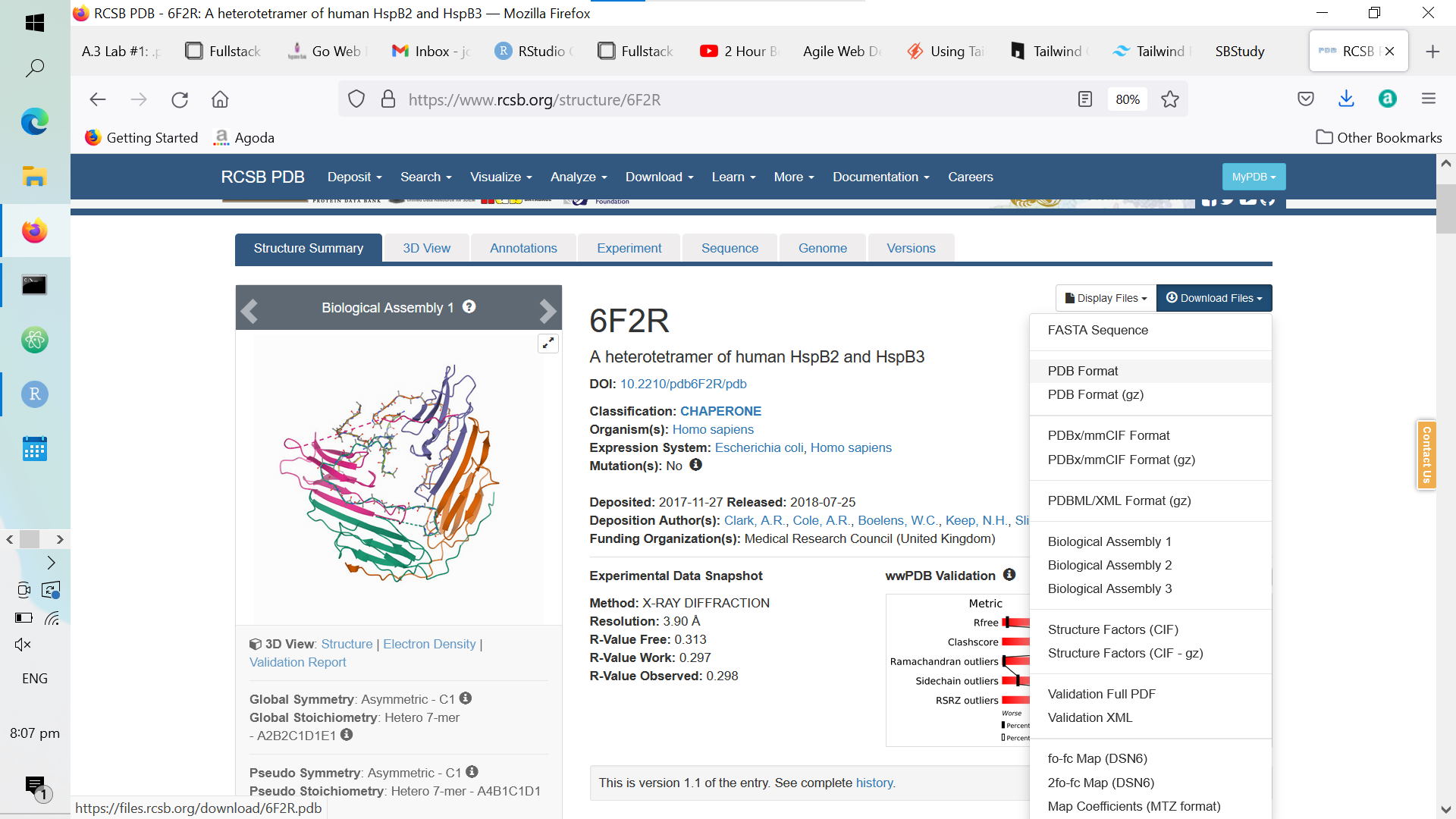
Figure A.4: Downloading a Random Protein’s .pdb File
Then, open up the .pdb file that you have downloaded and pull up the official 1996 documentation for .pdb files; for each section in your selected protein’s .pdb file, explain the purpose of each record and its format. You may look at my lab submission as a reference (but whatever you do, don’t plagiarize it).
A.4.2 Task #2: editing a .pdb file
In the second task, you are given a .pdb file that is purposely missing its three-letter amino acid residue codes in its ATOM records. The .pdb file is shown below:
1 2 3 4 5 6 7 8
12345678901234567890123456789012345678901234567890123456789012345678901234567890
ATOM 38 N A -33.869 10.617 7.317 1.00 76.26 N
ATOM 39 CA A -34.134 9.234 6.904 1.00 74.56 C
ATOM 40 C A -33.089 8.813 5.875 1.00 73.03 C
ATOM 41 O A -32.729 9.593 4.989 1.00 72.13 O
ATOM 42 CB A -35.552 9.101 6.317 1.00 75.48 C
ATOM 43 CG A -36.647 9.421 7.338 1.00 77.63 C
ATOM 44 OD1 A -36.667 8.874 8.452 1.00 81.93 O
ATOM 45 ND2 A -37.570 10.304 6.961 1.00 75.30 N
ATOM 46 N A -32.604 7.582 5.965 1.00 74.10 N
ATOM 47 CA A -31.570 7.168 5.019 1.00 77.48 C
ATOM 48 C A -32.043 6.300 3.846 1.00 73.71 C
ATOM 49 O A -31.538 5.201 3.613 1.00 76.65 O
ATOM 50 CB A -30.416 6.504 5.793 1.00 83.44 C
ATOM 51 CG A -29.901 7.343 6.934 1.00 90.57 C
ATOM 52 ND1 A -29.361 8.601 6.754 1.00 92.16 N
ATOM 53 CD2 A -29.899 7.125 8.275 1.00 92.50 C
ATOM 54 CE1 A -29.051 9.123 7.930 1.00 91.80 C
ATOM 55 NE2 A -29.369 8.247 8.871 1.00 92.32 N
ATOM 56 N A -33.001 6.831 3.089 1.00 67.38 N
ATOM 57 CA A -33.551 6.148 1.924 1.00 59.99 C
ATOM 58 C A -33.046 6.785 0.618 1.00 55.78 C
ATOM 59 O A -32.178 7.656 0.640 1.00 54.77 O
ATOM 60 CB A -35.079 6.191 1.993 1.00 58.20 C
ATOM 61 CG A -35.615 7.603 2.156 1.00 55.61 C
ATOM 62 OD1 A -36.782 7.745 2.593 1.00 55.40 O
ATOM 63 OD2 A -34.876 8.561 1.843 1.00 55.57 O
ATOM 64 N A -33.583 6.349 -0.515 1.00 54.36 N
ATOM 65 CA A -33.167 6.876 -1.814 1.00 53.16 C
ATOM 66 C A -33.626 8.317 -2.061 1.00 51.53 C
ATOM 67 O A -33.041 9.039 -2.874 1.00 50.28 O
ATOM 68 CB A -33.673 5.962 -2.929 1.00 56.36 C
ATOM 69 CG A -33.144 4.523 -2.821 1.00 59.12 C
ATOM 70 CD A -33.329 3.750 -4.125 1.00 57.06 C
ATOM 71 CE A -32.718 2.350 -4.022 1.00 60.09 C
ATOM 72 NZ A -32.740 1.596 -5.327 1.00 61.46 N
ATOM 73 N A -34.685 8.721 -1.368 1.00 48.02 N
ATOM 74 CA A -35.197 10.077 -1.478 1.00 42.97 C
ATOM 75 C A -34.131 10.992 -0.891 1.00 43.60 C
ATOM 76 O A -33.858 12.059 -1.426 1.00 44.56 O
ATOM 77 CB A -36.487 10.260 -0.668 1.00 40.66 C
ATOM 78 CG1 A -37.593 9.371 -1.249 1.00 39.01 C
ATOM 79 CG2 A -36.885 11.737 -0.658 1.00 39.33 C
ATOM 80 CD1 A -38.855 9.309 -0.390 1.00 36.41 CNB: the column numbers on the first two lines have been provided for reference!
In this task, you will need to open the above .pdb file using PyMOL (or a suitable .pdb file viewer) and complete the following tasks:
- For columns 18 - 20, fill in the three-letter code for the amino acid that the atom belong sto.
- For column 26, number the ATOM records according to their amino acids starting from 1 (you can look at my lab to see how it’s done).
A.4.3 Task #3: creating a .pdb file
Using what you have learned about .pdb files so far, you will need to create a .pdb file for the structure of a water molecule.
The bond lengths of a water molecule are 0.957 Angstroms - the H-O-H bond angle is 109.5\(^\omicron\). Hence, first set the oxygen atom at the origin (0, 0, 0) using an ATOM record like so:
1 2 3 4 5 6 7
1234567890123456789012345678901234567890123456789012345678901234567890123456789
ATOM 1 O A 1 0.000 0.000 0.000 1.00 0.00 ONB: like the first .pdb example in task 2, the column numbers on the first few lines have been provided for reference!
Thereafter, add in the first hydrogen atom at the coordinates (0.957, 0, 0) using another ATOM record. It not necessary to use the aforementioned coordinates - (0, 0.957, 0) and (0, 0, 0.957) also work:
1 2 3 4 5 6 7
1234567890123456789012345678901234567890123456789012345678901234567890123456789
ATOM 1 O A 1 0.000 0.000 0.000 1.00 0.00 O
ATOM 2 H A 1 0.957 0.000 0.000 1.00 0.00 HWe can then perform some linear algebra to determine the coordinates of the final hydrogen atom.9
The matrix associated with the 2D, anti-clockwise rotation of points about an angle \(\theta\) is denoted as:
\[\begin{equation} \left[ \begin{matrix} \cos\theta & -\sin\theta \\ \sin\theta & \cos\theta \end{matrix} \right] \end{equation}\]
Hence, for an anti-clockwise rotation about the origin, the second hydrogen atom (using the first hydrogen atom’s coordinates as a reference) should be positioned at the following coordinates:
\[\begin{equation} \left[ \begin{matrix} \cos 109.5 & -\sin 109.5 \\ \sin 109.5 & \cos 109.5 \end{matrix} \right] * \left[ \begin{matrix} 0.957 \\ 0 \end{matrix} \right] \\ \approx \left[ \begin{matrix} -0.319 \\ 0.902 \end{matrix} \right] \end{equation}\]
NB: the third, z-coordinate has been purposely left out
Then, fill in the .pdb file using yet another ATOM record like such:
1 2 3 4 5 6 7
1234567890123456789012345678901234567890123456789012345678901234567890123456789
ATOM 1 O A 1 0.000 0.000 0.000 1.00 0.00 O
ATOM 2 H A 1 0.957 0.000 0.000 1.00 0.00 H
ATOM 3 H A 1 -0.319 0.902 0.000 1.00 0.00 HLastly, add in the TER and the END records:
1 2 3 4 5 6 7
1234567890123456789012345678901234567890123456789012345678901234567890123456789
ATOM 1 O A 1 0.000 0.000 0.000 1.00 0.00 O
ATOM 2 H A 1 0.957 0.000 0.000 1.00 0.00 H
ATOM 3 H A 1 -0.312 0.957 0.000 1.00 0.00 H
TER 4
END When you are done with this step, you will need to write a brief conclusion stating what you have learned and what you have taken away from this lab. You can look at my conclusion for reference.
It is not necessary to include the math behind the determination of the coordinates of the second hydrogen atom.↩︎