Chapter 4 Seurat QC Cell-level Filtering
library(Seurat)
library(data.table)
library(tidyverse)
library(magrittr)
library(gridExtra)4.1 Description
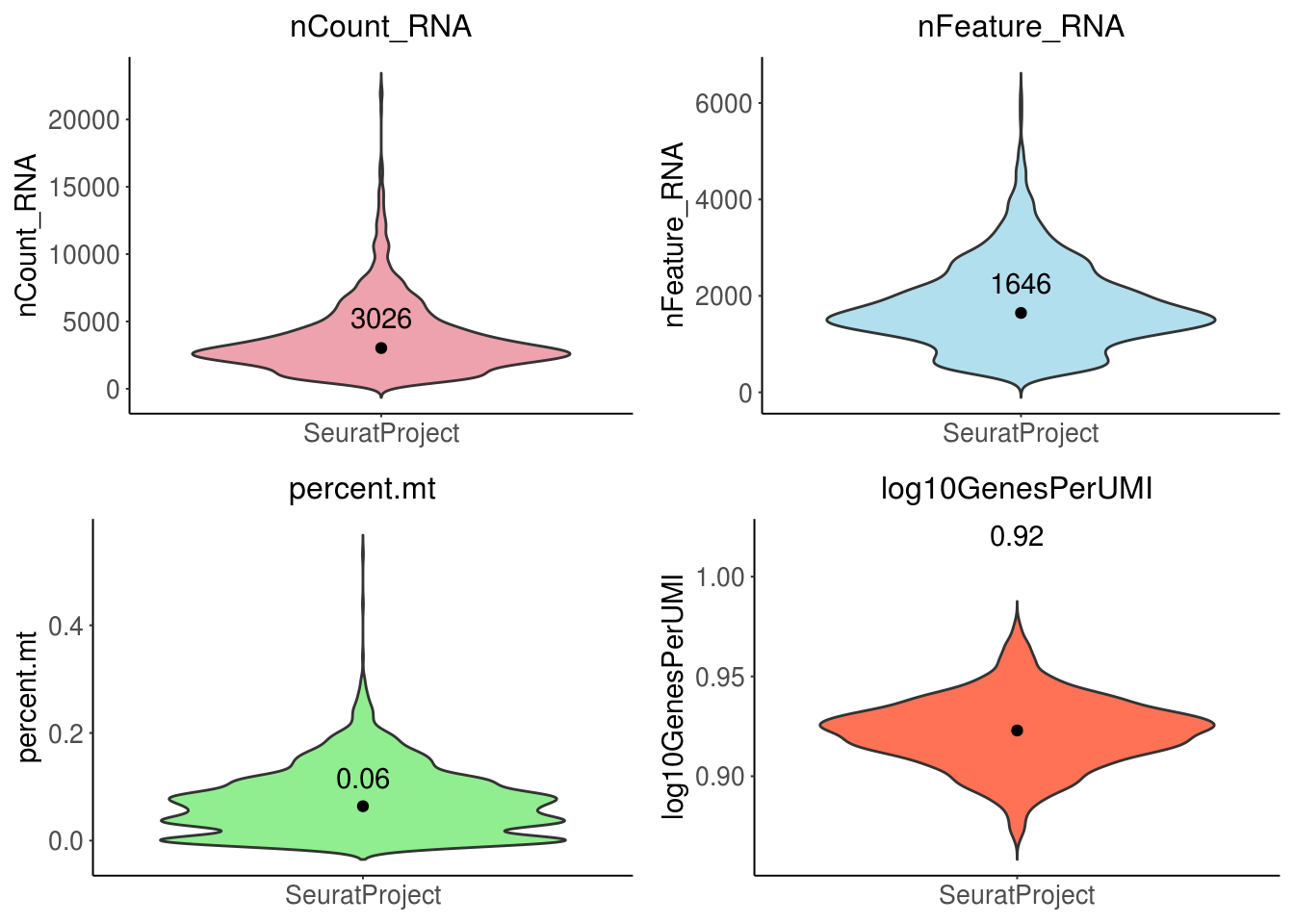
Basic quality control for snRNA-seq: check the distribution of
number of UMIs per cell
- should above 500
number of genes detected per cell
number of genes detected per UMI
- check the complexity. outlier cells might be cells have less complex RNA species like red blood cells. expected higher than 0.8
mitochondrial ratio
- dead or dying cells will cause large amount of mitochondrial contamination
4.2 Load seurat object
combined <- get(load('data/Demo_CombinedSeurat_SCT_Preprocess.RData'))4.3 Add other meta info
- fraction of reads mapping to mitochondrial gene
# for macaque, not all genes start with MT is mitochondrion genes
mt.gene <- c("MTARC2","MTFR1L","MTERF1","MTFR2","MTRF1L","MTRES1",
"MTO1","MTCH1","MTFMT","MTFR1","MTERF3","MTERF2","MTPAP",
"MTERF4","MTCH2",'MTIF2',"MTG2","MTIF3","MTRF1","MTCL1")
combined[["percent.mt"]] <- PercentageFeatureSet(combined, features = mt.gene )- number of genes detected per UMI
combined$log10GenesPerUMI <- log10(combined$nFeature_RNA) / log10(combined$nCount_RNA)4.4 Violin plots to check
- get the meta data
df <- as.data.table(combined@meta.data)
sel <- c("orig.ident", "nCount_RNA", "nFeature_RNA", "percent.mt", "log10GenesPerUMI")
df <- df[, sel, with = FALSE]
df[1:3, ]## orig.ident nCount_RNA nFeature_RNA percent.mt log10GenesPerUMI
## 1: SeuratProject 2740 1705 0.10795250 0.9400695
## 2: SeuratProject 3140 1687 0.09593860 0.9228424
## 3: SeuratProject 2539 1456 0.03738318 0.9290675- define plotting function
fontsize <- 10
linesize <- 0.35
gp.ls <- df[, 2:5] %>% imap( ~ {
# define lable fun
give.n <- function(x) {
return(c(y = median(x) + max(x) / 10, label = round(median(x), 2)))
}
# assign colors
col.ls <-
setNames(
c('lightpink2', 'lightblue2', 'lightgreen', 'coral1'),
c("nCount_RNA", "nFeature_RNA", "percent.mt", "log10GenesPerUMI")
)
ggplot(data = df, aes(x = orig.ident, y = .x)) +
geom_violin(trim = FALSE, fill = col.ls[.y]) +
ggtitle(label = .y) + ylab(label = .y) +
theme_bw() +
theme(
panel.grid.major = element_blank(),
panel.grid.minor = element_blank(),
strip.background = element_blank(),
panel.border = element_blank()
) +
theme(
axis.text = element_text(size = fontsize),
axis.line = element_line(colour = "black", size = linesize),
axis.ticks = element_line(size = linesize),
axis.title.x = element_blank(),
axis.ticks.length = unit(.05, "cm"),
plot.title = element_text(size = fontsize + 2, hjust = 0.5),
legend.position = 'none'
) +
stat_summary(fun = median, geom = "point", col = "black") + # Add points to plot
stat_summary(fun.data = give.n,
geom = "text",
col = "black")
})
grid.arrange(gp.ls[[1]], gp.ls[[2]], gp.ls[[3]], gp.ls[[4]], ncol = 2)