Chapter 9 diffBD nearest gene GO enrichment
9.1 Description
Go enrichment using clusterProfiler for nearest genes around lineag-specific CBSs
9.2 Load peak
## Section: load peaks
##################################################
file.ls <-
list.files('data/DiffBD/', full.names = T, pattern = "bed")
peaks <- list(#stable = file.ls[5],
liver = file.ls[2],
muscle = file.ls[3],
bcell = file.ls[1])9.3 Find genes
## Section: annotate
##################################################
#txdb <- TxDb.Mmusculus.UCSC.mm9.knownGene
txdb = TxDb.Hsapiens.UCSC.hg19.knownGene
promoter <-
getPromoters(TxDb = txdb,
upstream = 2000,
downstream = 2000)
peakAnnoList <- lapply(
peaks,
annotatePeak,
TxDb = txdb,
tssRegion = c(-3000, 3000),
verbose = FALSE
)
## Section: find genes
##################################################
genes <- lapply(peakAnnoList, function(i) as.data.frame(i)$geneId)
names(genes) = sub("_", "\n", names(genes))9.4 GO enrichment cluster
## Section: cluster enrichment
##################################################
compGO <- compareCluster(
geneCluster = genes,
fun = "enrichGO",
pvalueCutoff = 0.05,
pAdjustMethod = "BH",
OrgDb = org.Hs.eg.db,
ont = 'BP'
)9.5 GO enrichment per gene set
## Section: go enrichment
##################################################
go.ls <- genes %>% map( ~ {
res <-
enrichGO(
gene = .x,
ont = 'BP',
OrgDb = org.Hs.eg.db,
pvalueCutoff = 0.05,
pAdjustMethod = 'BH'
)
return(res)
})9.6 GO enrichment cluster barplot
g1 <- dotplot(compGO, showCategory = 5, title = "GO Enrichment Analysis")
g1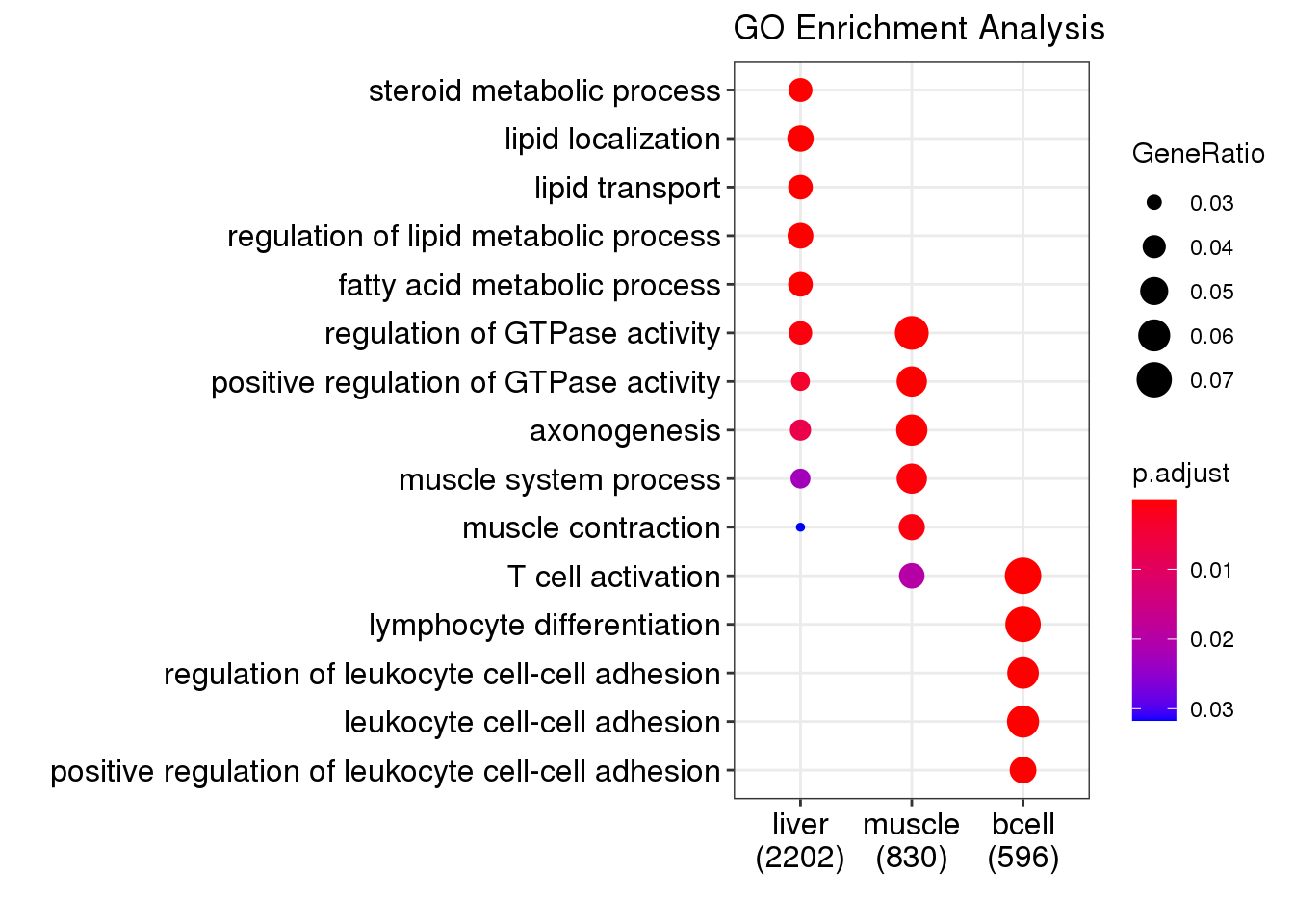
9.7 GO enrichment per set barplot
## Section: go term barplot
##################################################
barplotTerm <- function(object,
x = "Count",
color = 'p.adjust',
showCategory = 8,
font.size = 12,
title = "") {
## use *height* to satisy barplot generic definition
## actually here is an enrichResult object.
colorBy <- color
df <- fortify(object, showCategory = showCategory, by = x)
df$p.adjust <- -log10(df$p.adjust)
#df <- df[c(1:3,9:12,15,16),]
if (colorBy %in% colnames(df)) {
p <-
ggplot(df, aes_string(x = x, y = "Description", fill = colorBy)) +
theme_dose(font.size) +
scale_fill_continuous(
low = "red",
high = "blue",
name = color,
guide = guide_colorbar(reverse = TRUE)
)
} else {
p <- ggplot(df, aes_string(x = x, y = "Description")) +
theme_dose(font.size) +
theme(legend.position = "none")
}
p + geom_col(fill = color) + ggtitle(title) + xlab('-log10 FDR') + ylab(NULL)
}
bp.ls2 <- go.ls %>% imap(~ {
barplotTerm(
.x,
x = 'p.adjust',
color = 'blue',
showCategory = 10,
font.size = 4,
title = .y
)
})
do.call(cowplot::plot_grid, c(list(ncol = 3), bp.ls2))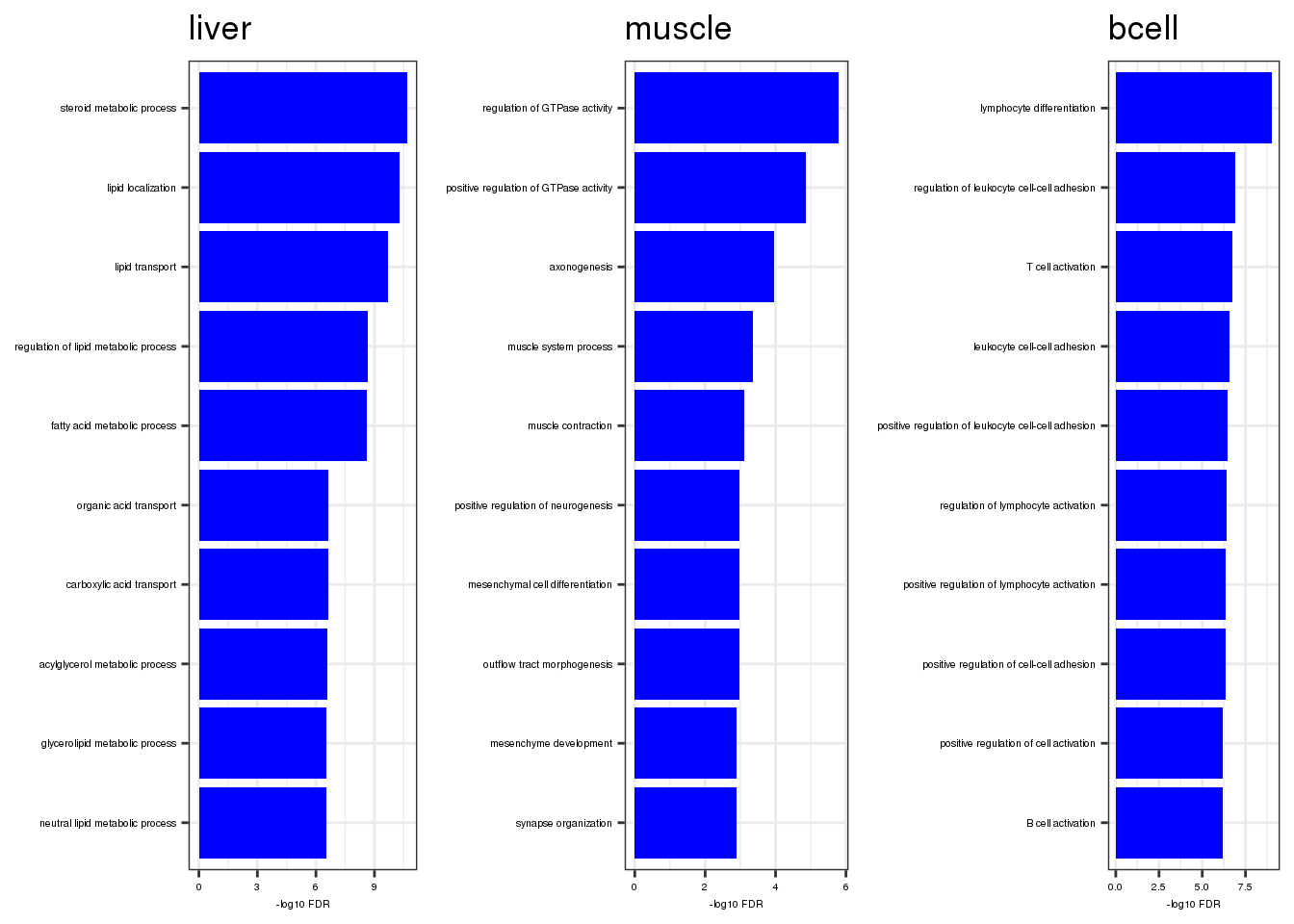
9.8 Save plots
ggsave("results/Genes/N9-Hep.Myo.Bcell-specificCBS-nearestgenes-function-goterm-dotplot.pdf", plot = g1,
width = 7.5, height = 5)
pdf('results/Genes/N9-Hep.Myo.Bcell-specificCBS-nearestgenes-function-goterm-barplot.pdf', width = 3*3, height = 3)
do.call(cowplot::plot_grid, c(list(ncol = 3), bp.ls2))
dev.off()