Chapter 10 diffBD genes expression
10.1 Description
check the expression levels for genes associated with lineage-specific CBS
10.2 Load expression mat
## Section: load expression table
##################################################
mat <- fread("~/lustrelyt/ChenQ/public/Human-Multitissue-CTCF/RNA-seq/Hep.Myo.Bcell_FPKM.table")10.3 Load lineage-specific/conserved CBS
## Section: load peaks
##################################################
file.ls <-
list.files('data/DiffBD/', full.names = T, pattern = "bed")
peaks <- list(
stable = file.ls[5],
liver = file.ls[2],
muscle = file.ls[3],
bcell = file.ls[1]
)10.4 Annotate genes for CBS
## Section: annotate genes by promoters
##################################################
txdb = TxDb.Hsapiens.UCSC.hg19.knownGene
gene.ls <- peaks %>% map( ~ {
peak <- readPeakFile(.x)
gene <-
seq2gene(
peak,
tssRegion = c(-1000, 1000),
flankDistance = 0,
TxDb = txdb
)
})10.5 Extract submat
## Section: subset fpkm table based on genes
##################################################
submat.ls <- gene.ls %>% map( ~ {
loci <- match(.x, mat$entrez)
loci <- loci[which(!is.na(loci))]
# subset fpkm table
submat <- mat[loci, 2:4]
# add gene symbol
gid <- mat$symbol[loci]
rownames(submat) <- make.names(gid, unique = TRUE)
# log norm
submat <- log2(submat + 1)
#submat <- t(scale(t(submat)))
return(submat)
})10.6 Boxplot to explore the expression levels across cells
stat_box_data <- function(y, upper_limit = quantile(df$value, 0.50) ) {
return( data.frame( y = upper_limit,
label = paste('count =', length(y), '\n','mean =', round(mean(y), 3), '\n')))
}
my_comparisons <- list(c('hepatocyte', 'myocyte'), c('hepatocyte', 'Bcell'), c('myocyte', 'Bcell'))
gp.ls <- submat.ls %>% imap(~ {
sample <- .y
submat <- .x
df <- melt(submat)
colnames(df) <- c('type', 'value')
df$type <- factor(df$type, levels = c('hepatocyte', 'myocyte', 'Bcell'))
df$value <- as.numeric(as.character(df$value))
fontsize = 10
linesize = 1
g <- ggboxplot(df, x = "type", y = "value",
color = 'type', size = .3, font.label = list(size = fontsize), outlier.shape = NA)+
ggtitle(sample) +
ylab('log2 FPKM')+ xlab(paste0("genes n=", nrow(submat) ))+
stat_summary(fun.data = stat_box_data, geom = "text", hjust = 0.5,vjust = 0.9)+
theme(legend.position = "none", axis.ticks.x = element_blank())+
stat_compare_means(comparisons = my_comparisons, label.y = c(6,7,8),method = 't.test', size = 2)+
scale_color_manual(values = c("hepatocyte" = rgb(223, 97, 20, maxColorValue = 255),
"myocyte" = rgb(66, 117, 224, maxColorValue = 255),
"Bcell" = rgb(125, 0, 250, maxColorValue = 255)))
return(g)
})
do.call(cowplot::plot_grid, c(list(ncol = 4), gp.ls))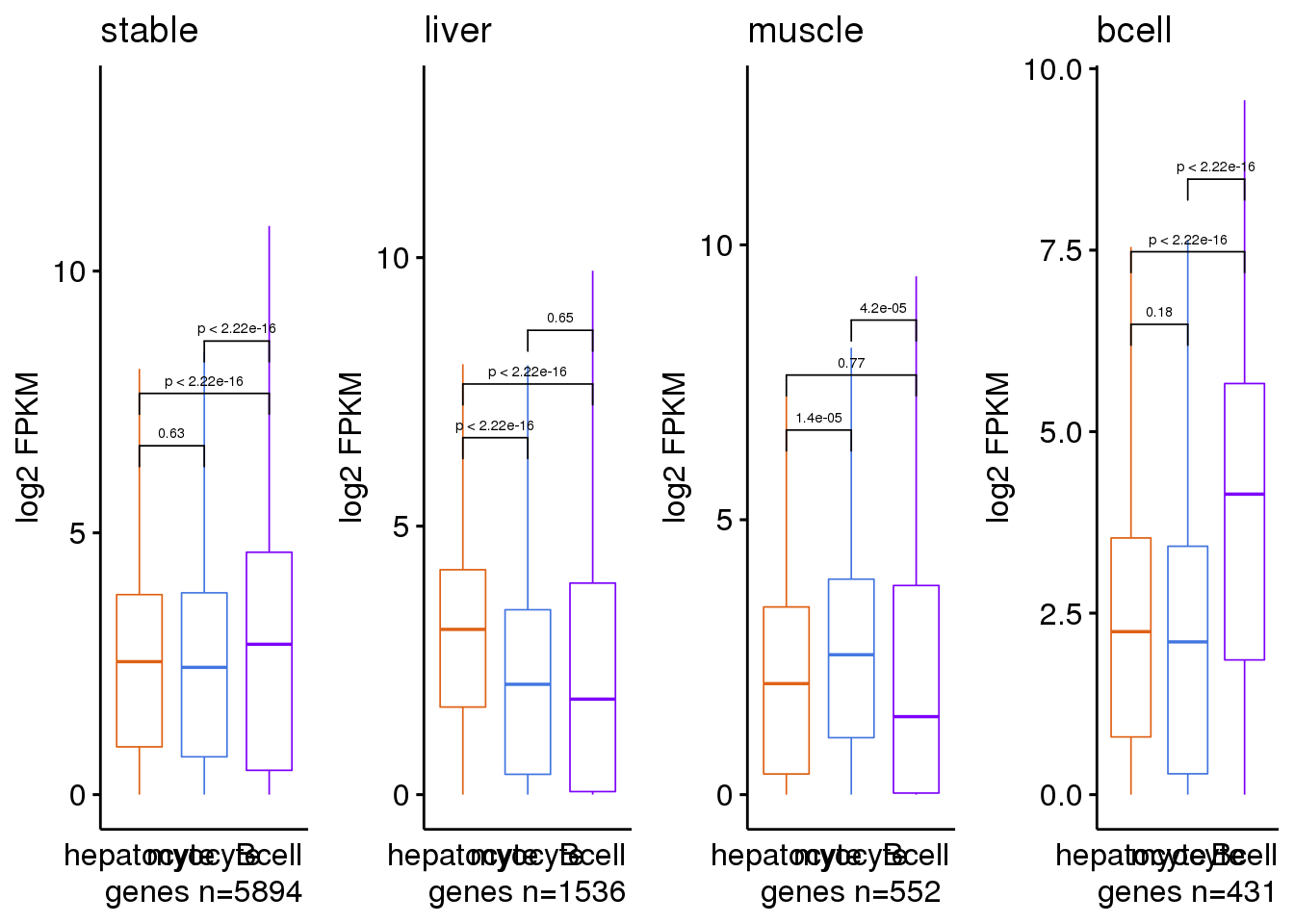
10.7 Save plots
pdf('results/Genes/N10-Hep.Myo.Bcell-specificCBS-AssociatedGenes-log2FPKM-fromENCODEData-boxplot.pdf',
width = 2.5*4, height = 3.5)
do.call(cowplot::plot_grid, c(list(ncol = 4), gp.ls))
dev.off()