Capítulo 7 Genética
7.1 Genética de populações
No estudo de genética de populações, alguns cálculos básicos são as frequências alélicas e genotípicas, verificar se está em equilíbrio de Hardy-Weinberg e também verificar o desequilíbrio de ligação.
Esses cálculos podem ser feitos com o pacote genetics (Warnes et al. 2021).
Vamos carregar alguns dados de exemplo:
## AMOSTRAS rs1800977 rs2230806 rs3764650 rs2279796 rs692383 rs3827225 grupo
## 1 AHC01 G/G C/C G/T A/G A/G G/G 1
## 2 AHC02 G/G C/T G/T G/G A/A A/G 1
## 3 AHC03 G/G T/T T/T A/G A/A G/G 1
## 4 AHC04 A/A C/C T/T G/G A/G A/G 1
## 5 AHC06 A/G C/C T/T A/A A/A A/A 1
## 6 AHC07 A/G C/C T/T A/G A/G A/A 1Vamos carregar o pacote genetics:
Agora vamos usar a função genotype, onde em sep informamos o separador de alelos nos dados.
## [1] "G/G" "G/G" "G/G" "A/A" "G/A" "G/A" "G/A" "G/A" "G/A" "G/G" "G/A" "G/G"
## [13] "G/A" "G/G" "G/G" "G/A" "G/G" "G/A" "G/A" "G/A" "G/A" "A/A" "G/G" "G/G"
## [25] "G/A" "G/G" "G/A" "G/A" "G/A" "G/A" "G/A" "G/G" "G/G" "G/G" "G/A" "G/G"
## [37] "G/A" "G/A" "G/G" "A/A" "G/G" "G/A" "G/A" "G/G" "G/G" "G/G" "G/A" "A/A"
## [49] "G/A" "G/A" "G/G" "G/G" "G/A" "A/A" "A/A" "G/G" "A/A" "G/G" "G/A" "G/G"
## [61] "A/A" "A/A" "G/A" "G/G" "G/A" "G/A" "G/A" "G/A" "G/G" "G/A" "G/G" "G/A"
## [73] "G/A" "G/G" "G/A" "G/A" "G/A" "G/A" "G/A" "G/A" "G/A" "G/G" "G/G" "G/G"
## [85] "G/G" "G/A" "A/A" "G/A" "G/A" "G/A" "G/G" "G/G" "G/G" "G/A" "G/A" "G/A"
## [97] "G/A" "G/G" "G/G" "G/A" "G/G" "G/G" "G/A" "G/A" "G/A" "G/G" "G/G" "G/G"
## [109] "G/G" "G/G" "G/A" "G/G" "G/A" "G/G" "G/A" "A/A" "A/A" "G/A" "G/A" "G/G"
## [121] "A/A" "G/A" "G/G" "G/A" "G/A" "G/G" "G/G" "G/A" "A/A" "A/A" "G/G" "G/A"
## [133] "G/A" "G/G" "G/G" "G/A" "G/G" "G/G" "G/A" "G/A" "G/G" "G/G" "G/A" "A/A"
## [145] "G/G" "G/A" "A/A" "G/G" "G/A" "G/G" "G/G" "G/G" "G/G" "G/A" "G/G" "G/A"
## [157] "G/G" "G/G" "G/A" "G/A" "G/G" "G/A" "G/G" "G/A" "G/G" "G/G" "G/G" "G/A"
## [169] "G/G" "G/A" "G/A" "G/A" "G/A" "G/A" "A/A" "G/A" "G/G" "G/A" "G/A" "G/A"
## [181] "G/G" "A/A" "G/A" "G/G" "G/A" "G/G" "G/A" "A/A" "G/A" "G/A" "G/A" "G/A"
## [193] "G/G" "G/A" "A/A" "G/G" "G/A" "G/G" "A/A" "G/A" "G/A" "G/A" "G/G" "G/G"
## [205] "G/A" "G/G" "G/G" "G/A" "G/G" "G/A" "G/A" "G/A" "G/A" "G/A" "G/A" "G/A"
## [217] "G/G" "G/A" "G/A" "A/A" "G/G" "G/G" "G/A" "G/G" "G/G" "G/G" "G/A" "G/A"
## [229] "G/A" "G/A" "G/G" "G/A" "G/G" "G/A" "G/G" "G/G" "G/A" "G/G" "G/G" "G/A"
## [241] "G/G" "G/A" "G/A" "G/A" "G/G" "G/G" "G/G" "G/G" "G/G" "G/A" "G/A" "G/A"
## [253] "G/A" "A/A" "G/G" "G/A" "G/G" "G/A" "G/A" "G/A" "G/A" "G/A" "G/A" "G/A"
## [265] "G/A" "G/A" "G/G" "G/A" "G/G" "G/G" "G/G" "G/G" "A/A" "G/G" "G/G" "G/G"
## [277] "G/A" "G/G" "G/G" "G/A" "G/A" "G/G" "G/G" "G/A" "A/A" "G/A" "A/A" "G/A"
## [289] "G/G" "G/A" "G/G" "G/A" "G/G" "G/A" "A/A" "A/A" "G/A" "G/G" "G/G" "G/A"
## [301] "G/G" "G/G" "G/G" "G/G" "G/A" "G/G" "G/G" "G/A" "G/A" "G/A" "G/G" "G/A"
## [313] "G/G" "G/G" "G/G" "G/G" "G/A" "G/A" "G/A" "G/A" "G/G" "G/G" "G/A" "G/A"
## [325] "A/A"
## Alleles: G APodemos ver os resultados de frequências usando summary:
##
## Number of samples typed: 325 (100%)
##
## Allele Frequency: (2 alleles)
## Count Proportion
## G 430 0.66
## A 220 0.34
##
##
## Genotype Frequency:
## Count Proportion
## G/G 135 0.42
## G/A 160 0.49
## A/A 30 0.09
##
## Heterozygosity (Hu) = 0.4485007
## Poly. Inf. Content = 0.3475435Para verificar o equilíbrio de Hardy-Weinberg usamos a função HWE.chisq:
##
## Pearson's Chi-squared test with simulated p-value (based on 10000
## replicates)
##
## data: tab
## X-squared = 3.2089, df = NA, p-value = 0.08419Podemos salvar os resultados em um arquivo txt, onde vamos usar o objeto snp para especificar de qual SNP são os resultados:
write("Frequencias alelicas e genotipicas.", file="res.genes.txt",append=T)
write(snp, file="res.genes.txt", append=T)
capture.output(resultado1, file="res.genes.txt",append = T)
capture.output(eqhw1, file="res.genes.txt", append = T)Também podemos gerar figuras com as frequências:


Para salvar as figuras com as frequências, onde filename=paste(snp,“alelo.jpg”, sep=““: serve para criar o nome do arquivo juntando o conteúdo de snp + alelo.jpg, sem espaço, definido por sep sem nada:
jpeg(filename=paste(snp,"alelo.jpg", sep=""))
plot(genotipos1, type="allele", what="percentage")
dev.off()## png
## 2jpeg(filename=paste(snp,"genotipo.jpg", sep=""))
plot(genotipos1, type="genotype", what="percentage")
dev.off()## png
## 2Desequilíbrio de ligação
Também podemos calcular o desequilíbrio de ligação entre 2 SNPs com a função LD, mas primeiro criamos os objetos com os dados dos SNPs com a função genotype:
rs1800977<-na.omit(genotype(dados$rs1800977, sep="/"))
rs2230806<-na.omit(genotype(dados$rs2230806, sep="/"))
ld<-LD(rs1800977,rs2230806)
ld##
## Pairwise LD
## -----------
## D D' Corr
## Estimates: -0.01494381 0.146423 -0.06881552
##
## X^2 P-value N
## LD Test: 3.078125 0.07935198 3257.2 Genotipagem com genetics e loop
Vamos juntar os conhecimentos de loops, que já aprendemos, para genotipar vários SNPs e criar um arquivo de saída mais elaborado:
No arquivo que estamos usando, tem uma coluna grupo (caso e controle). Usaremos essa coluna para criar 2 objetos:
Precisamos detalhar o que tem em resultados1:
## [1] "allele.names" "allele.freq" "genotype.freq" "Hu"
## [5] "pic" "n.typed" "n.total" "nallele"Em allele.fre e genotype.freq temos uma matriz com os resultados:
## Count Proportion
## G 430 0.6615385
## A 220 0.3384615## Count Proportion
## G/G 135 0.41538462
## G/A 160 0.49230769
## A/A 30 0.09230769Com base nisso, precisamos definir quais dados queremos salvar e criar uma tabela com as variáveis. Aqui criamos o objeto tabela.saida, que é um data.frame e especificamos as colunas:
tabela.saida<-data.frame(grupo=as.character(NA),SNP=as.character(NA),genot1=as.character(NA), N1=NA, F1=NA,genot2=as.character(NA), N2=NA, F2=NA,genot3=as.character(NA),N3=NA,F3=NA,alelo1=as.character(NA),N4=NA,F4=NA,alelo2=as.character(NA),N5=NA,F5=NA,hw=NA,qui_genot=NA,qui_alel=NA,stringsAsFactors = F)Agora vamos criar o loop, usando for, para fazer a genotipagem e equilíbrio de HW, de cada SNP, no grupo caso e no grupo controle, depois compare por qui-quadrado as frequências alélicas e genotípica entre casos e controles.
nomes<-colnames(dados) #nomes dos SNPs
i<-2 #número da coluna inicial para genotipar
snp<-7 # número da coluna final para genotipar
j<-1
for (i in i:snp) {
nome_snp<-nomes[i]
genotipos1<-na.omit(genotype(caso[,i], sep="/"))#genotipagem
resultado1<-summary(genotipos1)#sumariza resultados
eqhw1<-HWE.chisq(genotipos1)#equilíbrio de HW
nome_genot<-rownames(resultado1$genotype.freq)
nome_alelos<-rownames(resultado1$allele.freq)
tabela.saida[j,]<-data.frame(grupo=as.character("caso"),SNP=as.character(nome_snp),genot1=as.character(nome_genot[1]), N1=resultado1$genotype.freq[1,1], F1=resultado1$genotype.freq[1,2],genot2=as.character(nome_genot[2]), N2=resultado1$genotype.freq[2,1],F2=resultado1$genotype.freq[2,2], genot3=as.character(nome_genot[3]),N3=resultado1$genotype.freq[3,1],F3=resultado1$genotype.freq[3,2],alelo1=as.character(nome_alelos[1]),N4=resultado1$allele.freq[1,1],F4=resultado1$allele.freq[1,2],alelo2=as.character(nome_alelos[2]),N5=resultado1$allele.freq[2,1],F5=resultado1$allele.freq[2,2],hw=eqhw1$p.value,qui_genot=NA,qui_alel=NA,stringsAsFactors = F)
j<-j+1
genotipos2<-na.omit(genotype(controle[,i], sep="/"))#genotipagem
resultado2<-summary(genotipos2)#sumariza resultados
eqhw2<-HWE.chisq(genotipos2)#equilíbrio de HW
#qui-quadrado alelo
a<-resultado1$allele.freq[1,1]
b<-resultado1$allele.freq[2,1]
c<-resultado2$allele.freq[1,1]
d<-resultado2$allele.freq[2,1]
quadrado<-rbind(c(a,c),c(b,d))
quadrado
res_qui<-chisq.test(quadrado,correct=F)
#qui-quadrado genotipo
g<-resultado1$genotype.freq[1,1]
h<-resultado1$genotype.freq[2,1]
l<-resultado1$genotype.freq[3,1]
k<-resultado2$genotype.freq[1,1]
m<-resultado2$genotype.freq[2,1]
o<-resultado2$genotype.freq[3,1]
quadrado2<-rbind(c(g,k),c(h,m),c(l,o))
res_qui2<-chisq.test(quadrado2,correct=F)
tabela.saida[j,]<-data.frame(grupo=as.character("controle"),SNP=as.character(nome_snp),genot1=as.character(nome_genot[1]), N1=resultado2$genotype.freq[1,1], F1=resultado2$genotype.freq[1,2],genot2=as.character(nome_genot[2]), N2=resultado2$genotype.freq[2,1],F2=resultado2$genotype.freq[2,2], genot3=as.character(nome_genot[3]),N3=resultado2$genotype.freq[3,1],F3=resultado2$genotype.freq[3,2],alelo1=as.character(nome_alelos[1]),N4=resultado2$allele.freq[1,1],F4=resultado2$allele.freq[1,2],alelo2=as.character(nome_alelos[2]),N5=resultado2$allele.freq[2,1],F5=resultado2$allele.freq[2,2],hw=eqhw2$p.value,qui_genot=res_qui2$p.value,qui_alel=res_qui$p.value,stringsAsFactors = F)
i<-(i+1)
j<-j+1
}Verificando os resultados:
grupo | SNP | genot1 | N1 | F1 | genot2 | N2 | F2 | genot3 | N3 | F3 | alelo1 | N4 | F4 | alelo2 | N5 | F5 | hw | qui_genot | qui_alel |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
caso | rs1800977 | G/G | 79 | 0.4030612 | G/A | 96 | 0.4897959 | A/A | 21 | 0.10714286 | G | 254 | 0.6479592 | A | 138 | 0.3520408 | 0.3501650 | ||
controle | rs1800977 | G/G | 56 | 0.4341085 | G/A | 64 | 0.4961240 | A/A | 9 | 0.06976744 | G | 176 | 0.6821705 | A | 82 | 0.3178295 | 0.1065893 | 0.5055572 | 0.3671365 |
caso | rs2230806 | C/C | 91 | 0.4642857 | C/T | 90 | 0.4591837 | T/T | 15 | 0.07653061 | C | 272 | 0.6938776 | T | 120 | 0.3061224 | 0.3173683 | ||
controle | rs2230806 | C/C | 67 | 0.5193798 | C/T | 48 | 0.3720930 | T/T | 14 | 0.10852713 | C | 182 | 0.7054264 | T | 76 | 0.2945736 | 0.2920708 | 0.2505753 | 0.7535973 |
caso | rs3764650 | T/T | 135 | 0.7758621 | T/G | 36 | 0.2068966 | G/G | 3 | 0.01724138 | T | 306 | 0.8793103 | G | 42 | 0.1206897 | 1.0000000 | ||
controle | rs3764650 | T/T | 79 | 0.8061224 | T/G | 16 | 0.1632653 | G/G | 3 | 0.03061224 | T | 174 | 0.8877551 | G | 22 | 0.1122449 | 0.1042896 | 0.5473538 | 0.7691475 |
caso | rs2279796 | A/A | 55 | 0.2777778 | A/G | 91 | 0.4595960 | G/G | 52 | 0.26262626 | A | 201 | 0.5075758 | G | 195 | 0.4924242 | 0.2571743 | ||
controle | rs2279796 | A/A | 31 | 0.2460317 | A/G | 69 | 0.5476190 | G/G | 26 | 0.20634921 | A | 131 | 0.5198413 | G | 121 | 0.4801587 | 0.2820718 | 0.2845827 | 0.7607357 |
caso | rs692383 | A/A | 83 | 0.4234694 | A/G | 85 | 0.4336735 | G/G | 28 | 0.14285714 | A | 251 | 0.6403061 | G | 141 | 0.3596939 | 0.4345565 | ||
controle | rs692383 | A/A | 53 | 0.4140625 | A/G | 59 | 0.4609375 | G/G | 16 | 0.12500000 | A | 165 | 0.6445312 | G | 91 | 0.3554688 | 1.0000000 | 0.8488922 | 0.9126687 |
caso | rs3827225 | G/G | 103 | 0.5282051 | G/A | 76 | 0.3897436 | A/A | 16 | 0.08205128 | G | 282 | 0.7230769 | A | 108 | 0.2769231 | 0.7174283 | ||
controle | rs3827225 | G/G | 74 | 0.5692308 | G/A | 49 | 0.3769231 | A/A | 7 | 0.05384615 | G | 197 | 0.7576923 | A | 63 | 0.2423077 | 0.6277372 | 0.5623675 | 0.3261360 |
Para terminar vamos salvar os resultados em um arquivo xlsx:
7.3 Heredogramas
Também podemos gerar heredogramas usando o R, para isso usamos o pacote kinship2 (Sinnwell and Therneau 2022) e carregar o arquivo de exemplo:
## ids dad mom sex dead caract
## 1 1 NA NA 1 0 0
## 2 2 NA NA 2 1 0
## 3 3 NA NA 1 0 0
## 4 4 1 2 2 0 0
## 5 5 1 2 1 0 0
## 6 6 NA NA 2 0 0Nessa planilha temos uma coluna com a identificação única de de cada indivíduo (ids), duas colunas identificando os pais (dad e mom), uma coluna para o sexo, uma coluna com a informação se o indivíduo está vivo ou não (dead) e uma última coluna (caract) que indica se o indivíduo tem a característica de interesse.
Usando a função pedigree:
family <- pedigree(id=familia$ids, dadid=familia$dad, momid=familia$mom, affected=familia$caract, sex=familia$sex, status=familia$dead)E plotando o heredograma:
plot.pedigree(family,cex = 0.8, col = c("red","blue","green","red","blue","green","red","blue","green","black"))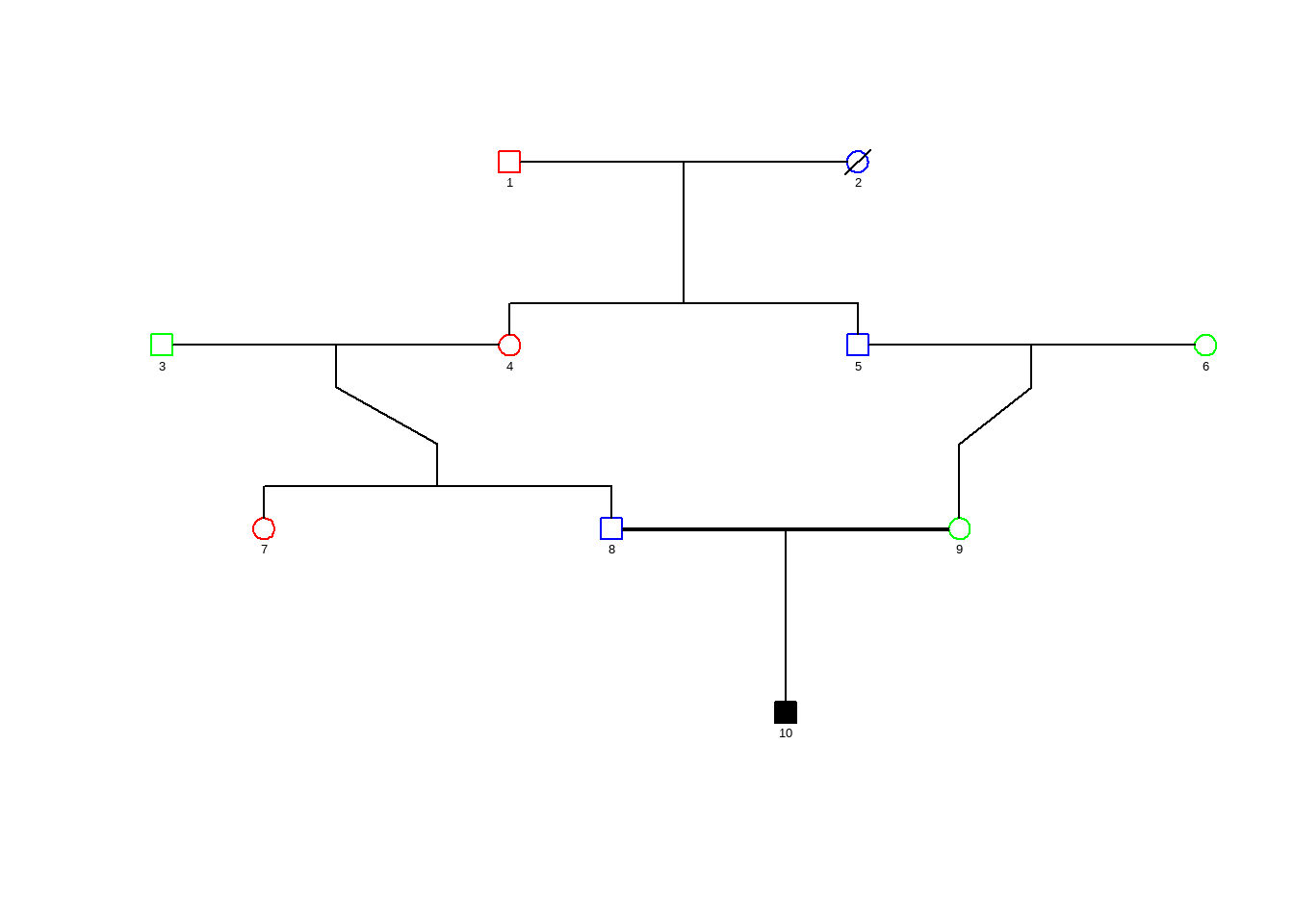
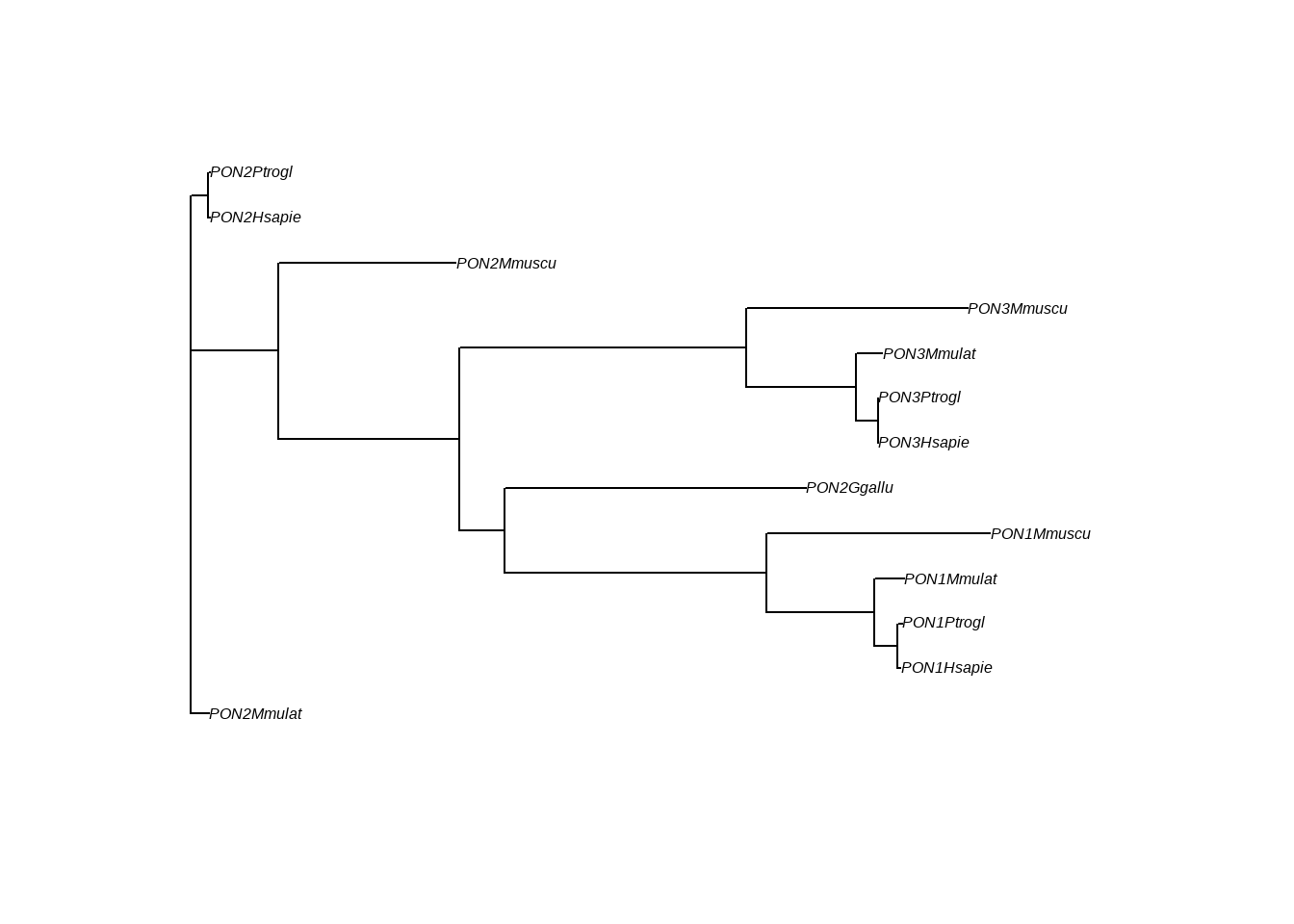
7.4 Análises filogenéticas
O pacote APE (Analysis of Phylogenetics and Evolution) (paradis2019?) permite fazer vários tipos de análises (http://ape-package.ird.fr/).
Podemos pegar sequências de DNA diretamente do Genbank:
library(ape)
id_seq<-c("NM_001081850.1","NM_000055.3","NM_009738.3")
seq_bche<-read.GenBank(id_seq)
seq_bche## 3 DNA sequences in binary format stored in a list.
##
## Mean sequence length: 3495.333
## Shortest sequence: 1809
## Longest sequence: 6209
##
## Labels:
## NM_001081850.1
## NM_000055.3
## NM_009738.3
##
## Base composition:
## a c g t
## 0.315 0.192 0.194 0.299
## (Total: 10.49 kb)Também é possível ler um arquivo fasta:
## 13 DNA sequences in binary format stored in a list.
##
## All sequences of same length: 1065
##
## Labels:
## PON1Hsapie
## PON1Ptrogl
## PON1Mmulat
## PON1Mmuscu
## PON2Hsapie
## PON2Ptrogl
## ...
##
## Base composition:
## a c g t
## 0.284 0.215 0.228 0.274
## (Total: 13.85 kb)E com a função nj podemos fazer uma árvore pelo método neighbor-joining (Trees 1987).