Chapter 2 Introduction to R
Nhan Thi Ho, MD, PhD
2.1 What is R?
R is a free software environment for statistical computing and graphics. To download, install, and learn more, go to R cran (R Core Team 2024b).
2.2 RStudio
RStudio is an integrated development environment (IDE) for R. It includes a console, syntax-highlighting editor that supports direct code execution, as well as tools for plotting, history, debugging and workspace management. To download, install, and learn more, go to RStudio page.
2.3 Rmarkdown
R Markdown is a file format for making dynamic documents with R.
It turns your analyses into high quality REPRODUCIBLE documents, reports, presentations and dashboards (Allaire et al. 2024).
Learn more at Rmarkdown page.
2.4 Base R Example
R comes with a few base packages for base functions.
Example: calculate, generate, summary data:
## [1] 6## [1] 3## [1] 5#Check how to use a function
#?rnorm
#use the function
data1<- rnorm(n=30,mean=1.5, sd=0.5)
summary(data1)## Min. 1st Qu. Median Mean 3rd Qu. Max.
## 0.6084 1.2695 1.5829 1.5306 1.8650 2.4256## Min. 1st Qu. Median Mean 3rd Qu. Max.
## 0.369 1.345 2.021 1.978 2.426 4.643Basic statistical test: t-test example:
##
## Welch Two Sample t-test
##
## data: data1 and data2
## t = -2.4155, df = 53.351, p-value = 0.01917
## alternative hypothesis: true difference in means is not equal to 0
## 95 percent confidence interval:
## -0.81885042 -0.07595964
## sample estimates:
## mean of x mean of y
## 1.530573 1.9779782.5 Install, load, update R packages
In RStudio: packages>install>packagename From R console: type: install.packages(“packagename”)
Install and load 01 package, which is my R package “metamicrobiomeR” (N. Ho 2020):
Install and load multiple packages
#install multiple packages
Packages <- c("knitr","rmarkdown","installr","haven","readr","yaml","tidyverse")
#install.packages(Packages)
#load multiple packages
lapply(Packages, library, character.only = TRUE)Commands useful for updating packages
# download and install newest version of the package
#update.packages()
# shows which packages have updates
#old.packages ()
# looks for new packages that are not already installed
#new.packages ()Getting help for package, function, data
2.6 Working with R objects
2.6.1 Vector
A vector is a collection of elements:
## [1] 1 3 4 7 8 10## [1] "red" "blue" "white"## [1] 1 3 5 7 9## [1] "boring" "boring" "boring"2.6.2 Matrix
A matrix is a 2-dimensional representation of a vector (two-dimensional rectangular data set):
## [,1] [,2] [,3]
## [1,] 1 4 8
## [2,] 3 7 102.6.3 Array
An array is an n dimensional representation of a vector:
## , , 1
##
## [,1] [,2] [,3]
## [1,] 1 3 4
##
## , , 2
##
## [,1] [,2] [,3]
## [1,] 7 8 102.6.4 List
A list is a collection of “bins”, each containing any kind of R object:
## [[1]]
## [1] 1 3 4 7 8 10
##
## [[2]]
## [,1] [,2] [,3]
## [1,] 1 4 8
## [2,] 3 7 10
##
## [[3]]
## , , 1
##
## [,1] [,2] [,3]
## [1,] 1 3 4
##
## , , 2
##
## [,1] [,2] [,3]
## [1,] 7 8 10
##
##
## [[4]]
## [1] "red" "blue" "white"2.6.5 Data frame
A data frame is a list in tabular form where each “bin” contains a data vector of the same length. A data frame is the usual tabular data set familiar to epidemiologists. Each row is an record and each column (“bin”) is a field. Both matrix and data.frame are tabular data-types.
- All columns in a matrix must have the same data type (numeric, character, etc.) and the same length.
- A data frame is more general than a matrix, in that different columns can have different modes (numeric, character, factor, etc.). Just like a table in a database or excel sheet.
smoker <- c("Y", "Y", "N", "N", "Y", "Y", "Y", "N", "Y")
cancer <- c("Y", "N", "N", "Y", "Y", "Y", "N", "N", "Y")
quant<-c(1,2,3,4,5,6,7,8,9)
mydf <- data.frame(smoker, cancer,quant);
mydf[1:4,] # display rows 1 to 4## smoker cancer quant
## 1 Y Y 1
## 2 Y N 2
## 3 N N 3
## 4 N Y 42.6.6 Assessing the structure of data objects
## 'data.frame': 701 obs. of 23 variables:
## $ id : chr "k__bacteria.p__actinobacteria" "k__bacteria.p__bacteroidetes" "k__bacteria.p__firmicutes" "k__bacteria.p__fusobacteria" ...
## $ Estimate.genderMale : num 0.2269 -0.1549 -0.056 -0.0648 -0.0887 ...
## $ Std. Error.genderMale : num 0.121 0.119 0.104 0.282 0.118 ...
## $ t value.genderMale : num 1.874 -1.305 -0.537 -0.23 -0.751 ...
## $ Pr(>|t|).genderMale : num 0.0619 0.1931 0.5919 0.8186 0.4532 ...
## $ Estimate.bfNon_exclusiveBF : num -0.3688 0.2557 0.2362 -0.0605 0.3699 ...
## $ Std. Error.bfNon_exclusiveBF : num 0.139 0.136 0.116 0.364 0.134 ...
## $ t value.bfNon_exclusiveBF : num -2.647 1.887 2.035 -0.166 2.764 ...
## $ Pr(>|t|).bfNon_exclusiveBF : num 0.00857 0.0602 0.04276 0.86812 0.00606 ...
## $ Estimate.bfNo_BF : num -1.329 0.135 0.356 -0.403 1.153 ...
## $ Std. Error.bfNo_BF : num 0.412 0.499 0.392 1.083 0.407 ...
## $ t value.bfNo_BF : num -3.223 0.27 0.908 -0.372 2.834 ...
## $ Pr(>|t|).bfNo_BF : num 0.00141 0.7873 0.36478 0.7103 0.0049 ...
## $ Estimate.age.sample : num 0.1251 0.0242 0.0115 0.0285 -0.1348 ...
## $ Std. Error.age.sample : num 0.0377 0.0367 0.0319 0.0857 0.038 ...
## $ t value.age.sample : num 3.318 0.66 0.359 0.333 -3.549 ...
## $ Pr(>|t|).age.sample : num 0.001022 0.510068 0.719525 0.73934 0.000448 ...
## $ pval.adjust.genderMale : num 0.48 0.899 0.961 0.961 0.961 ...
## $ pval.adjust.bfNon_exclusiveBF: num 0.059 0.2333 0.1767 0.9976 0.0448 ...
## $ pval.adjust.bfNo_BF : num 0.0219 1 1 1 0.0468 ...
## $ pval.adjust.sample : num 0.00786 0.82595 0.85375 0.85375 0.00505 ...
## $ study : chr "Subramanian et al 2014 (Bangladesh)" "Subramanian et al 2014 (Bangladesh)" "Subramanian et al 2014 (Bangladesh)" "Subramanian et al 2014 (Bangladesh)" ...
## $ pop : chr "Bangladesh" "Bangladesh" "Bangladesh" "Bangladesh" ...2.6.7 Useful functions to assess R objects
| Function | Returns |
|---|---|
| str | summary of data object structure |
| attributes | list with data object attributes |
| mode | mode of object |
| typeof | type of object; similar to mode but includes double and integer, if applicable |
| length | length of object |
| class | class of object, if it exists |
| dim | vector with object dimensions, if applicable |
| nrow | number of rows, if applicable |
| ncol | |
| dimnames | list containing vectors of names for each dimension, if applicable |
| rownames | vector of row names of a matrix-like object |
| colnames | vector of column names of a matrix-like object |
| names | vector of names for the list (for a data frame returns field names) |
| row.names | vector of row names for a data frame |
2.6.8 Understanding vectors
Operation of vectors
## [1] 50 100 150## [1] 50 100 1502.6.8.1 Boolean queries and subsetting of vectors
## [1] "Nhan" "Hien" "Phuong" "Tien" "Duy"## [1] 41 39 33 26 25## [1] TRUE TRUE TRUE FALSE FALSEthirtysomething <- (ms.ages >= 30) & (ms.ages < 40)
lecturer[thirtysomething] # indexing using logical## [1] "Hien" "Phuong"## Nhan Hien Phuong Tien Duy
## 41 39 33 26 252.6.8.2 Operations on single vectors
Some operations on single vectors.
| Function | Description | Function | Description |
|---|---|---|---|
| sum | summation | rev | reverse order |
| cumsum | cumulative | sum | order order |
| diff | x[i+1]-x[i] | sort | sort |
| prod | product | rank | rank |
| cumprod | cumulative product | sample | random sample |
| mean | mean | quantile | percentile |
| median | median | var | variance, covariance |
| min | minimum | sd | standard deviation |
| max | maximum | table | tabulate character vector |
| range | range | xtabs | tabulate factor vector |
2.7 Managing biomedical data in R
2.7.1 Entering data
Enter simple data frame
subjname <- c('Lop', 'Chung', 'Minh')
subjno <- 1:length(subjname)
age <- c(34, 56, 56)
sex <- c('Male', 'Male', 'Female')
dat <- data.frame(subjno, subjname, age, sex); dat## subjno subjname age sex
## 1 1 Lop 34 Male
## 2 2 Chung 56 Male
## 3 3 Minh 56 Female2.7.2 Importing data from a file
From local: (Example data of Bangladeshi children (Subramanian et al. 2014)).
#your path to dataset
fdir1<-"./metamicrobiomeR-master/inst/extdata/QIIME_outputs/Bangladesh/"
#Read in text file
dat1<-read.delim(paste(fdir1,"Subramanian_et_al_mapping_file.txt",sep=""),header=T,sep ="")
#Or simply:
#dat1<-read.delim("./metamicrobiomeR-master/inst/extdata/QIIME_outputs/Bangladesh/Subramanian_et_al_mapping_file.txt",header=T,sep ="")From URL:
2.7.3 Some other data import functions
Data import with “rio” makes our life much easier! All you need is import()!
#install.packages("rio")
#install.packages(c('csvy', 'feather', 'fst', 'hexView', 'readODS', 'rmatio'))
library(rio)
test1<-import(paste(fdir1,"Subramanian_et_al_mapping_file.txt",sep=""))Import excel files with multiple sheets:
#import always get only 1 dataframe
test<-import(paste(fdir1,"nature13421-s2.xlsx",sep=""),sheet=1,skip=1)
#Import multiple sheets of an excel file and create a list of dataframes
list1<-import_list(paste(fdir1,"nature13421-s2.xlsx",sep=""))
#str(list1)
#View(list1[[1]])Multiple files from a directory
2.7.5 Basic data manipulation with “tidyverse”
“tidyverse” includes a collection of open source packages for the R programming language that share similar design philosophy, grammar, and data structures” for tidy data (Wickham 2023b).
2.7.5.1 Pipes
Pipes take the output of a command, and feed it as the input of another command.
library(tidyverse)
#pipe from library(magrittr), loaded along with other packages
9 %>% sqrt %>% `+`(8) ## [1] 112.7.5.3 Change column names
#change column name
colnames(dat2a)<-tolower(colnames(dat2a))
oldname<-colnames(dat2a)
newname<-c("child.id",
"family.id",
"cohort",
"gender",
"zygosity",
"whz","waz","haz",
"age.first.fe",
"age.last.fe",
"no.fe.sam",
"sam.int.mean",
"month.ebf",
"age.first.solid",
"no.diarhea.yr",
"pc.days.diarrhea",
"frac.sam.atb.7day",
"set")
dat2a<-dat2a %>% rename_at(vars(oldname), ~newname)
#colnames(dat2a)2.7.5.4 Subset data by row
dat.tw<-dat2a %>% filter(cohort == "Healthy Twins & Triplets")
#more filter
dat.tw<-dat2a %>%
filter(cohort == "Healthy Twins & Triplets")%>%
filter(!is.na(zygosity))%>%
filter(between(whz,-1,1))
#or
dat.tw<-dat2a %>%
filter(cohort == "Healthy Twins & Triplets" &
!is.na(zygosity) &
between(whz,-1,1)) 2.7.5.8 Editting data
## child.id family.id cohort gender zygosity
## Length:50 Length:50 Length:50 Length:50 Length:50
## Class :character Class :character Class :character Class :character Class :character
## Mode :character Mode :character Mode :character Mode :character Mode :character
##
##
##
## whz waz haz age.first.fe age.last.fe
## Min. :-1.73842 Min. :-3.7555 Min. :-4.6586 Min. : 1.00 Min. :286.0
## 1st Qu.:-0.88598 1st Qu.:-2.6037 1st Qu.:-3.4314 1st Qu.: 5.00 1st Qu.:455.0
## Median :-0.47063 Median :-1.5116 Median :-1.5749 Median : 6.00 Median :700.5
## Mean :-0.42223 Mean :-1.5148 Mean :-1.8570 Mean : 9.78 Mean :600.6
## 3rd Qu.:-0.03458 3rd Qu.:-0.7189 3rd Qu.:-0.7189 3rd Qu.: 9.75 3rd Qu.:706.0
## Max. : 1.20133 Max. : 1.6698 Max. : 1.6698 Max. :37.00 Max. :738.0
## no.fe.sam sam.int.mean month.ebf age.first.solid no.diarhea.yr pc.days.diarrhea
## Min. : 9.00 Min. :22.56 Min. :0.0000 Min. :1.216 Min. :0.000 Min. : 0.000
## 1st Qu.:18.00 1st Qu.:28.29 1st Qu.:0.1971 1st Qu.:4.920 1st Qu.:1.271 1st Qu.: 1.163
## Median :21.00 Median :30.48 Median :1.0348 Median :6.932 Median :2.916 Median : 2.742
## Mean :19.92 Mean :31.34 Mean :2.0716 Mean :6.690 Mean :2.971 Mean : 3.125
## 3rd Qu.:23.00 3rd Qu.:33.27 3rd Qu.:3.2523 3rd Qu.:8.361 3rd Qu.:4.054 3rd Qu.: 4.314
## Max. :26.00 Max. :42.29 Max. :7.3260 Max. :9.790 Max. :8.814 Max. :10.826
## frac.sam.atb.7day set
## Min. :0.00000 Length:50
## 1st Qu.:0.08083 Class :character
## Median :0.11882 Mode :character
## Mean :0.18328
## 3rd Qu.:0.28373
## Max. :0.500002.8 Basic data visualization
Below are some examples of basic data visualization with ggplot2 .
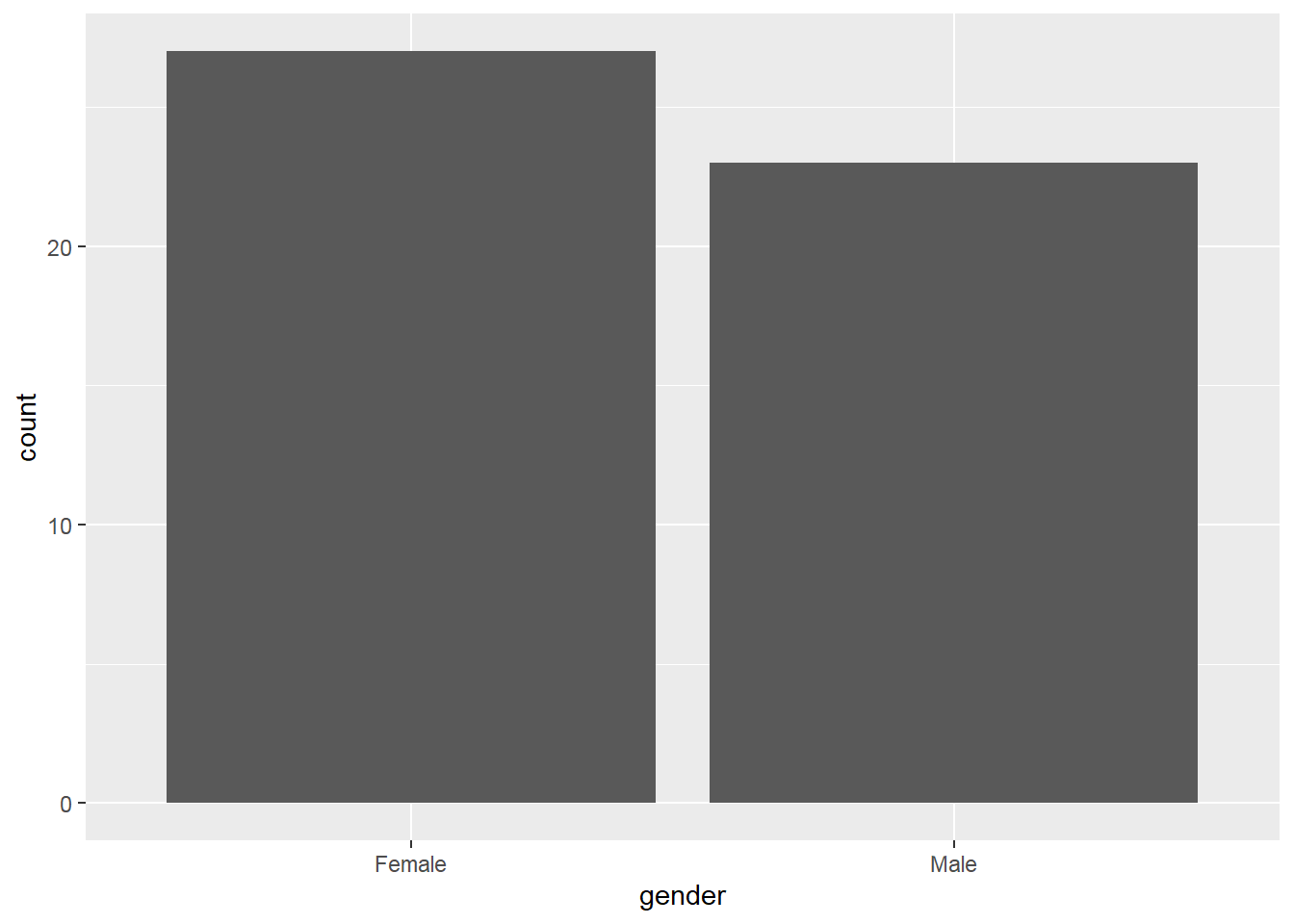
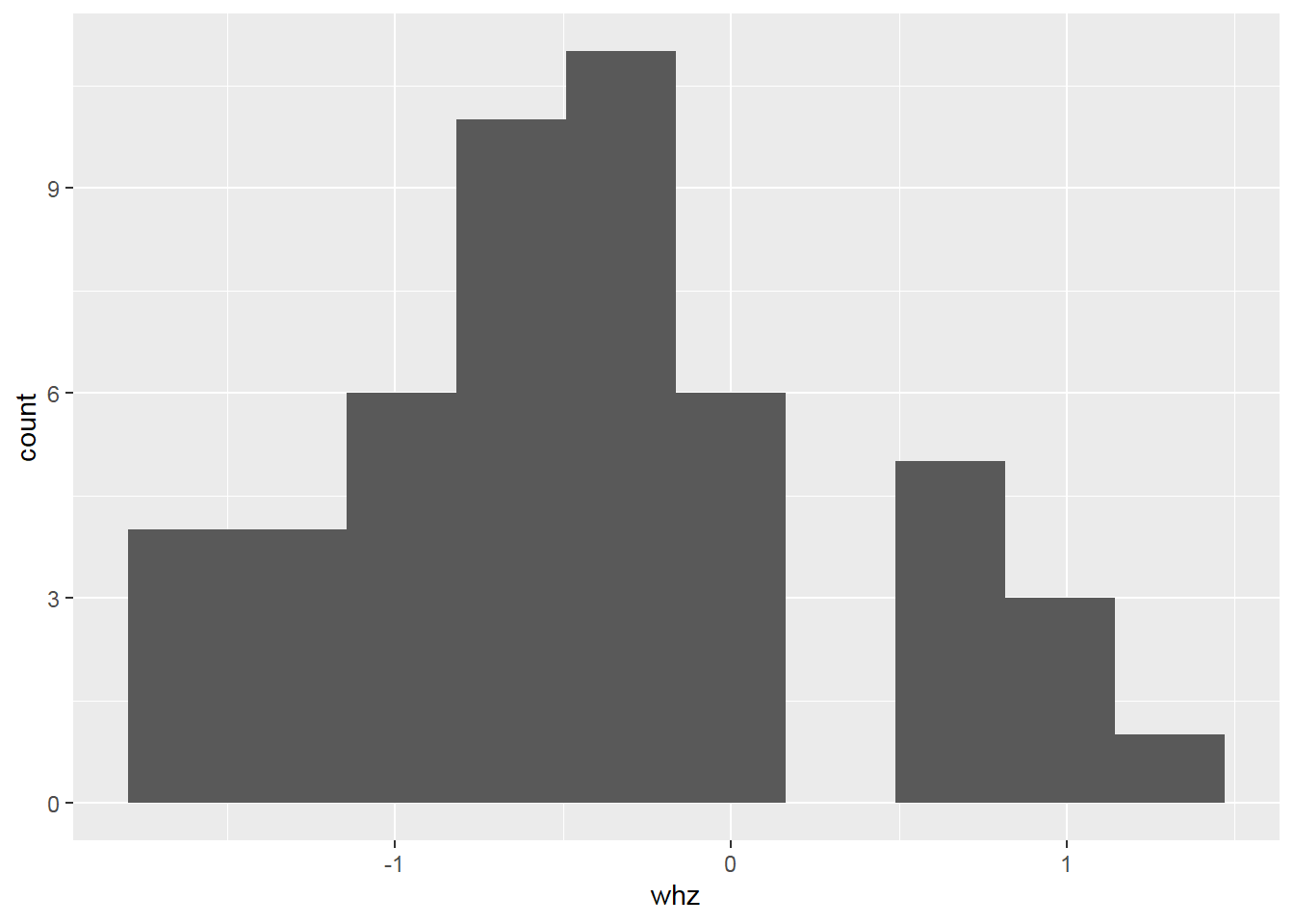
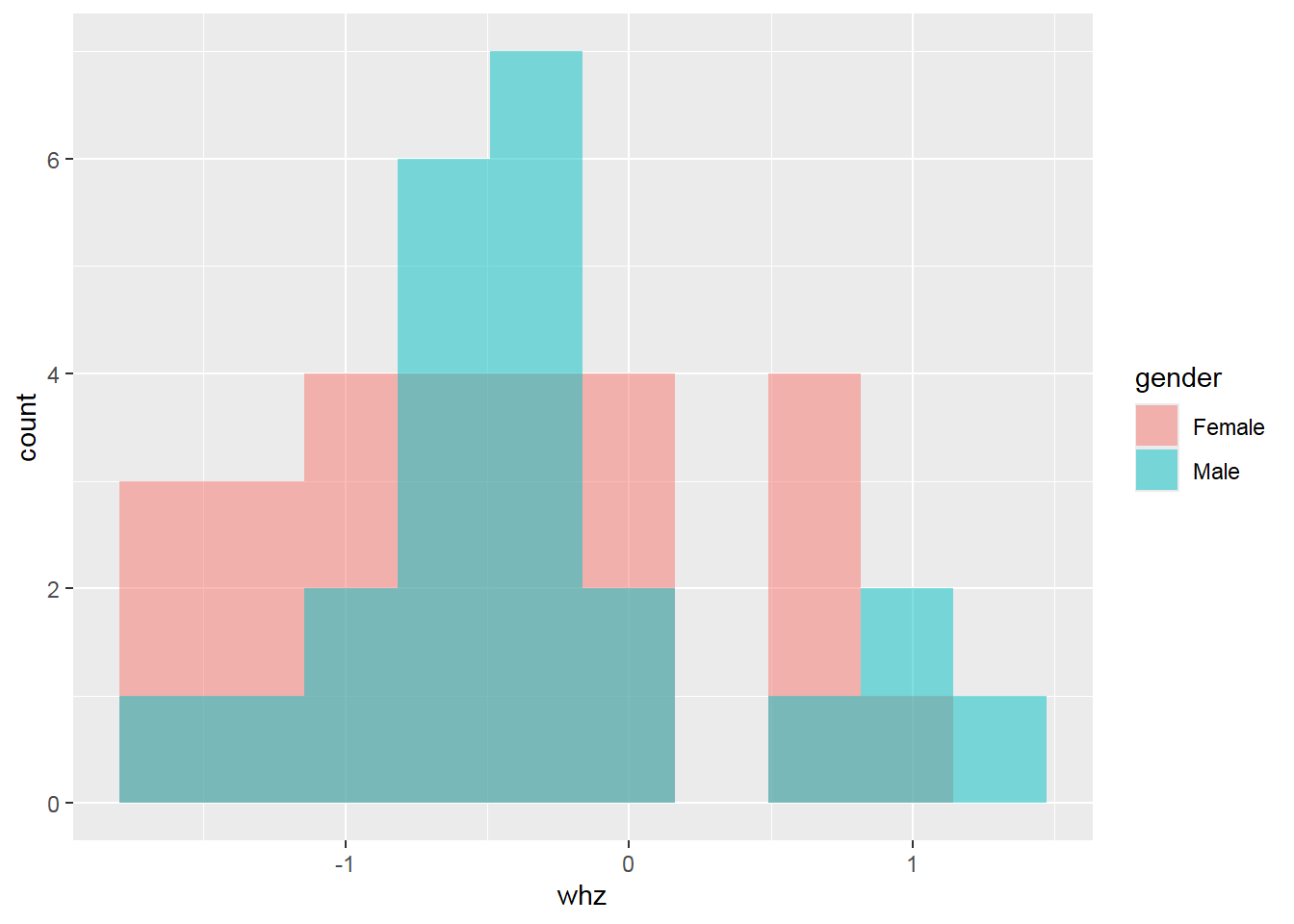
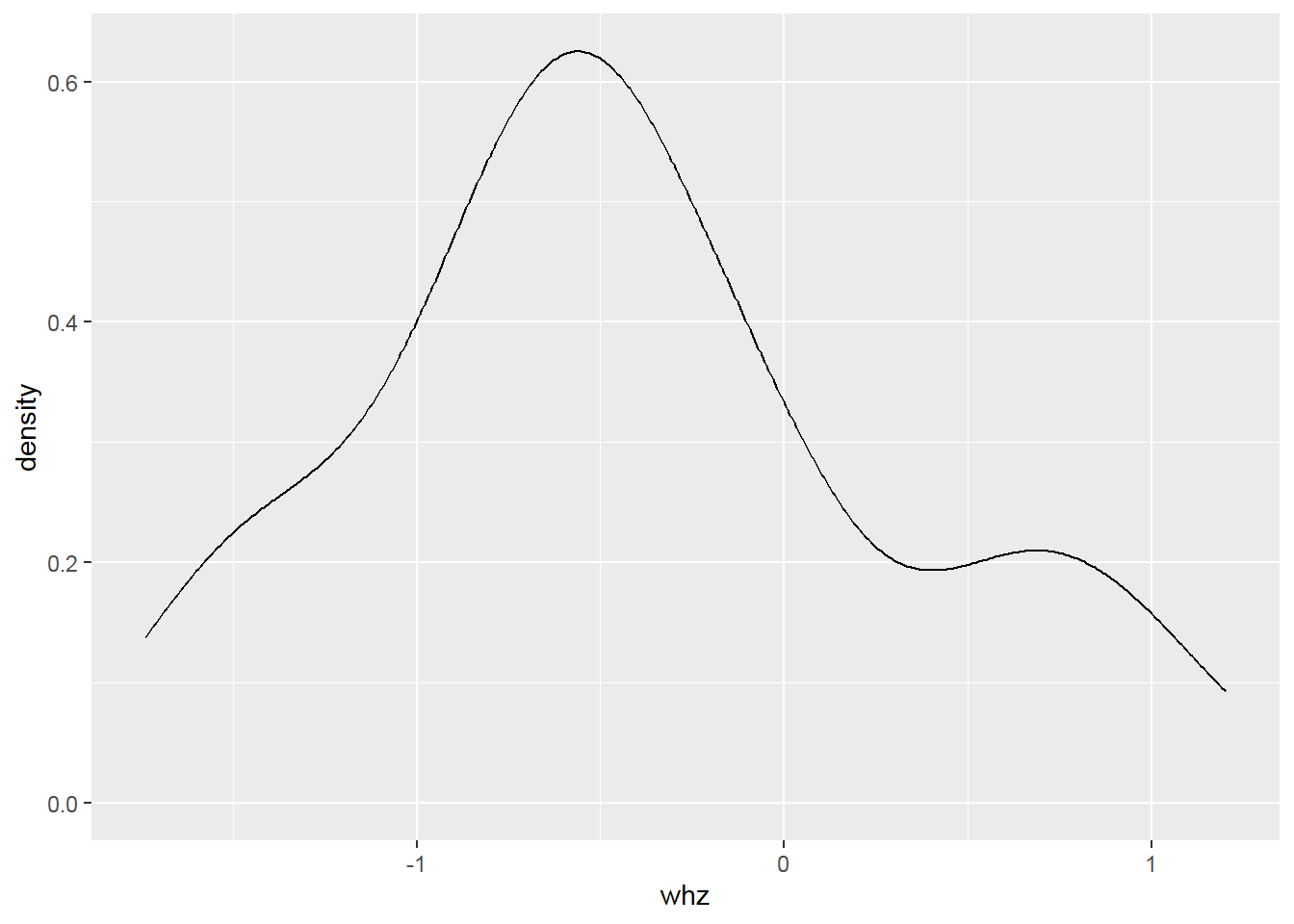
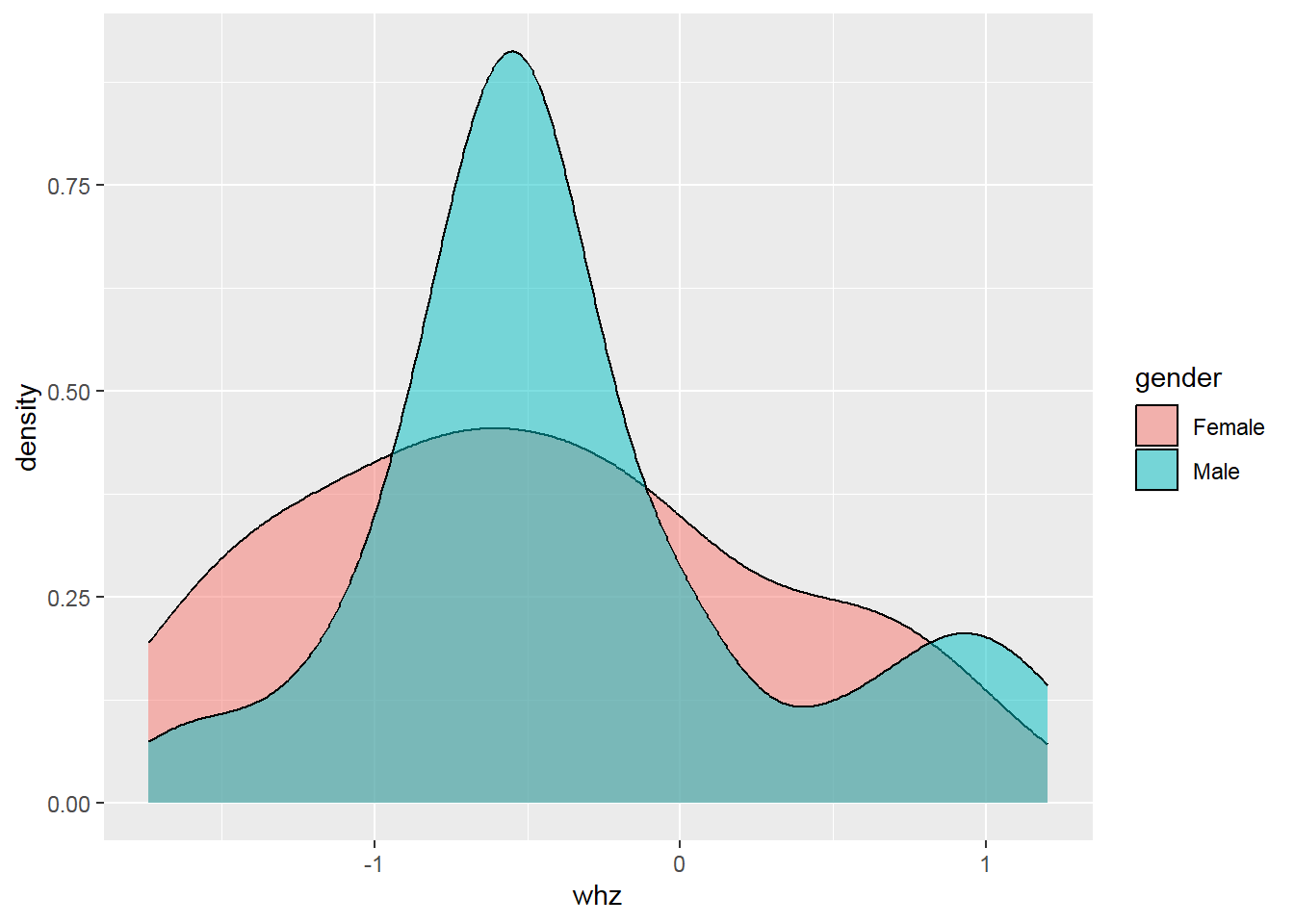
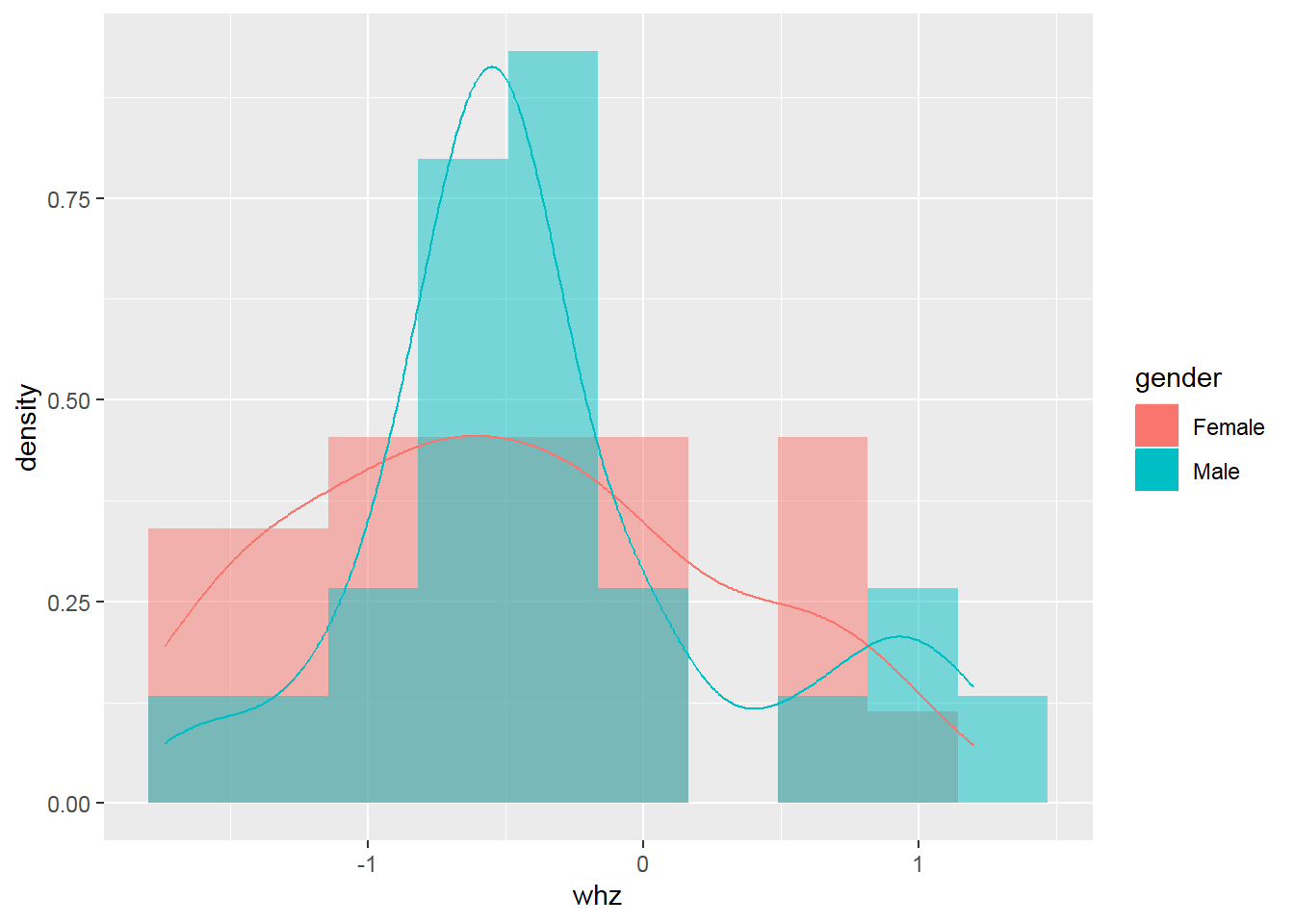
2.8.4 Histogram with density
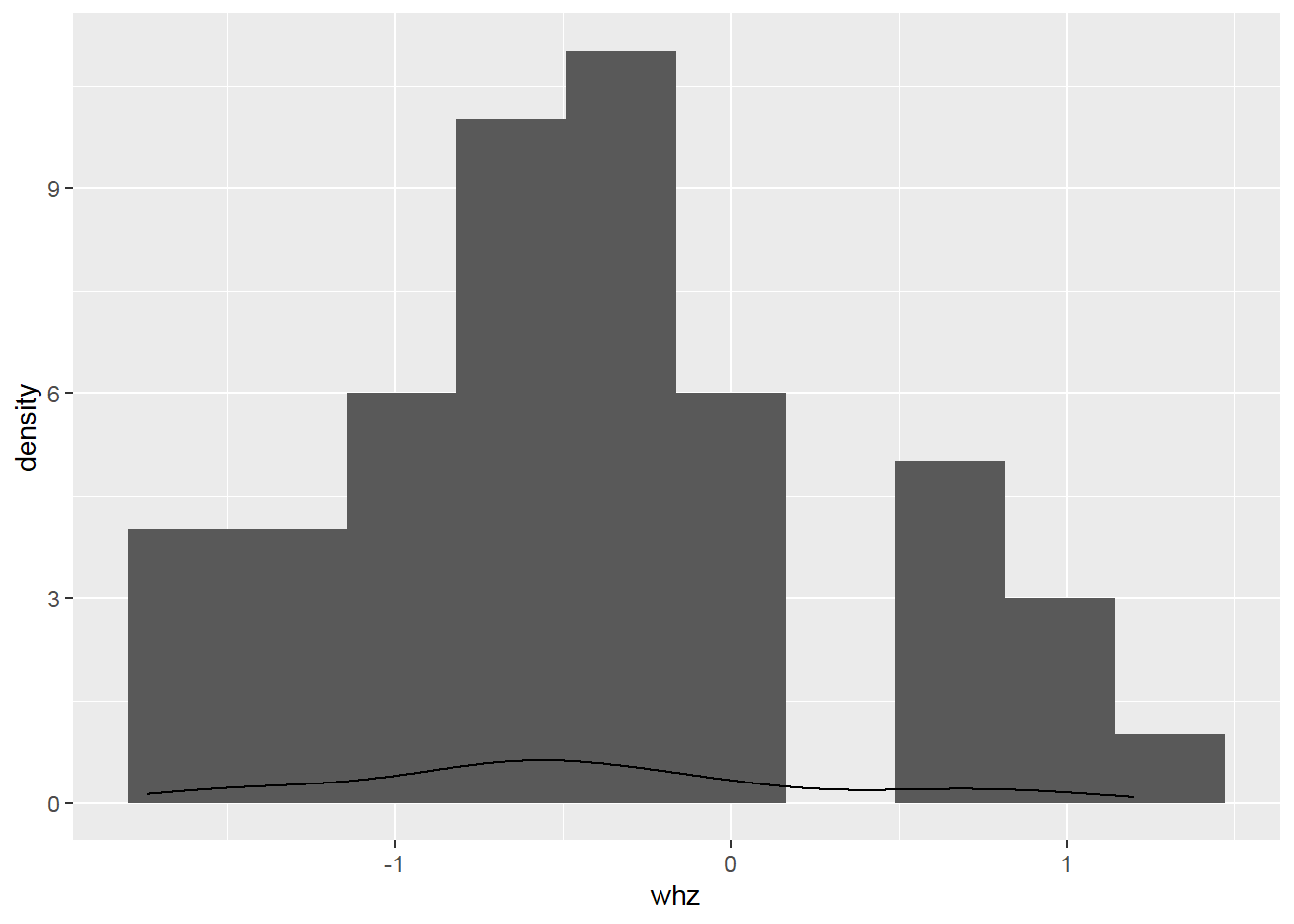
Something goes wrong! => Create a histogram with a density scale using the computed variable ..density..:
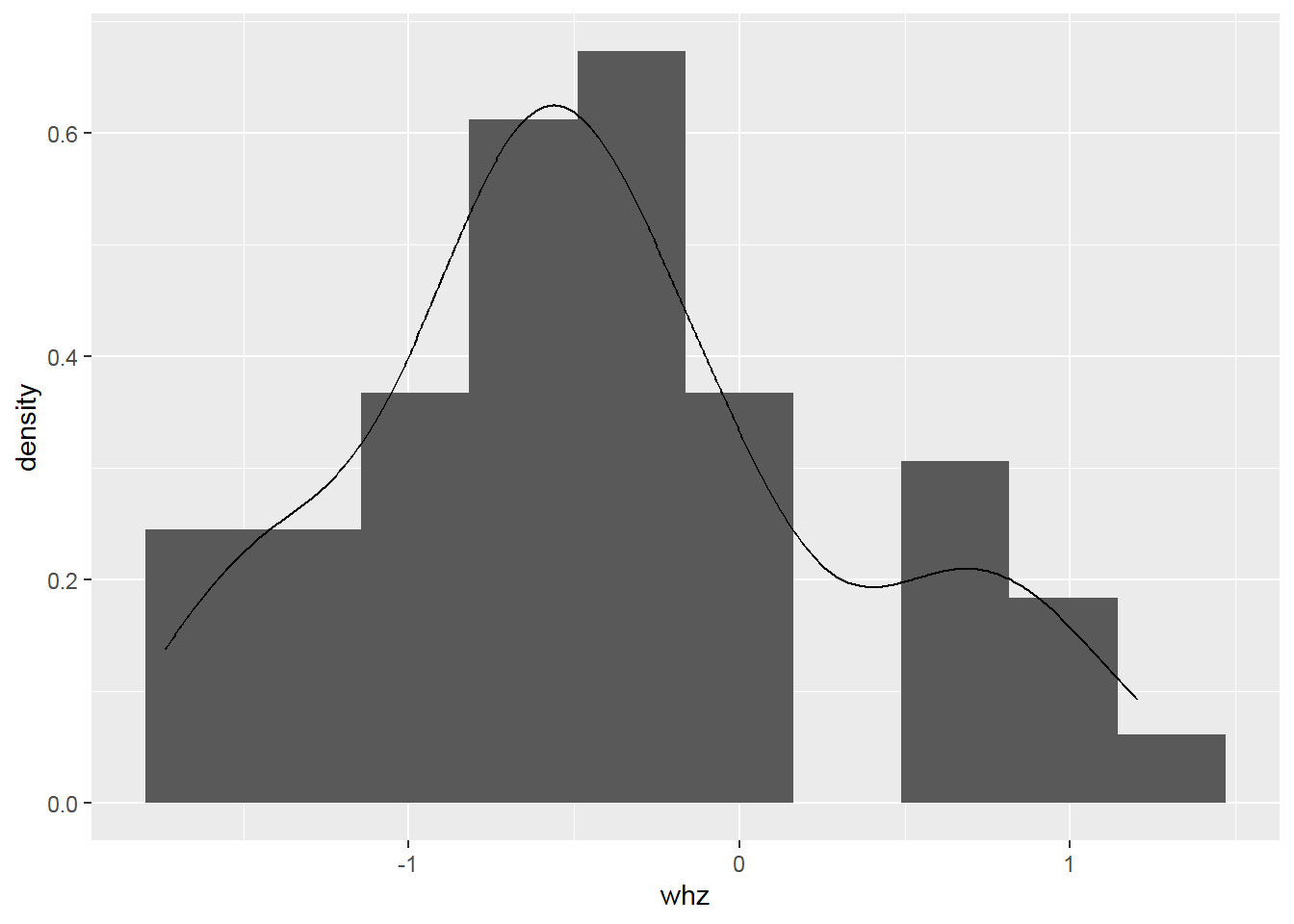
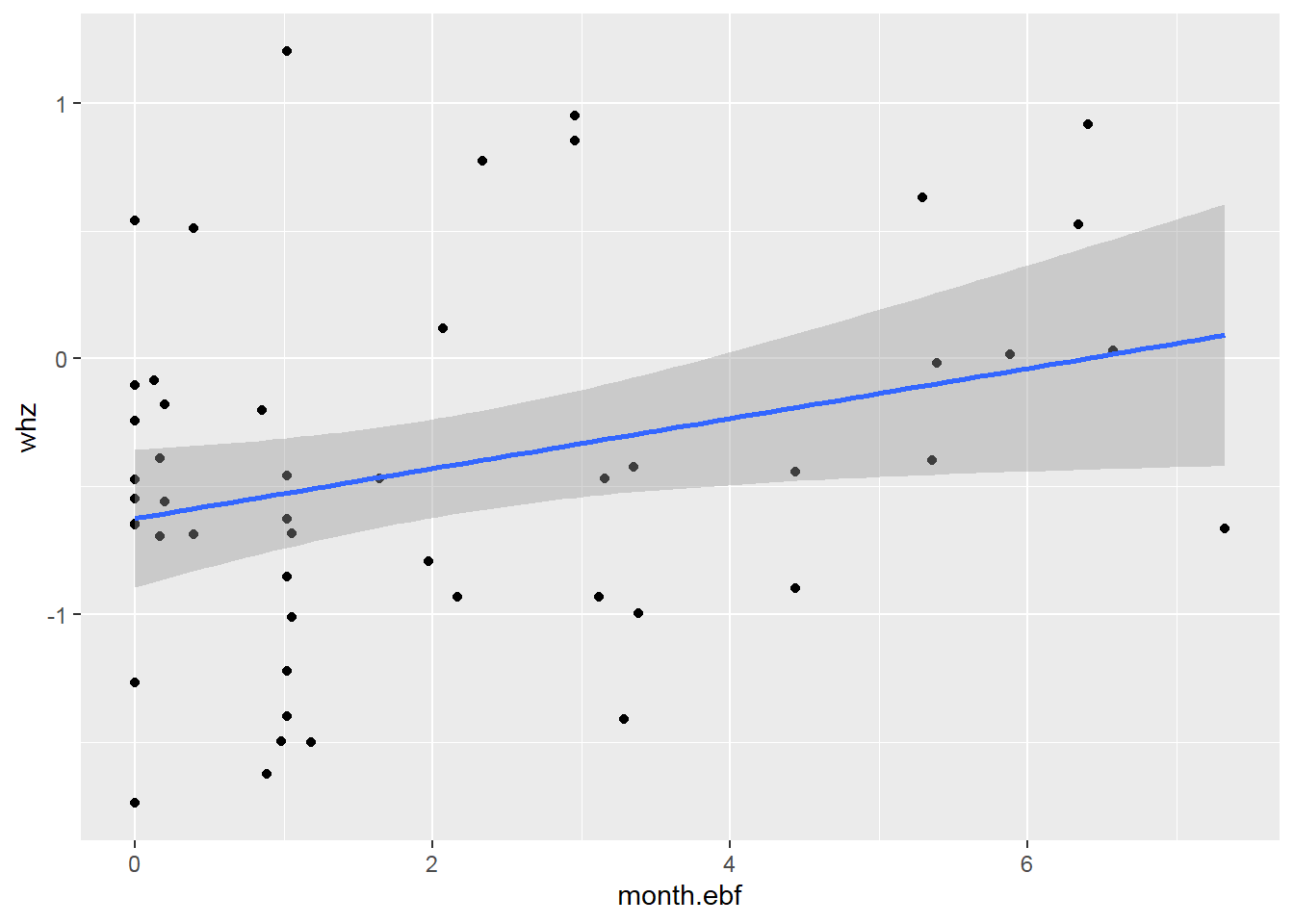
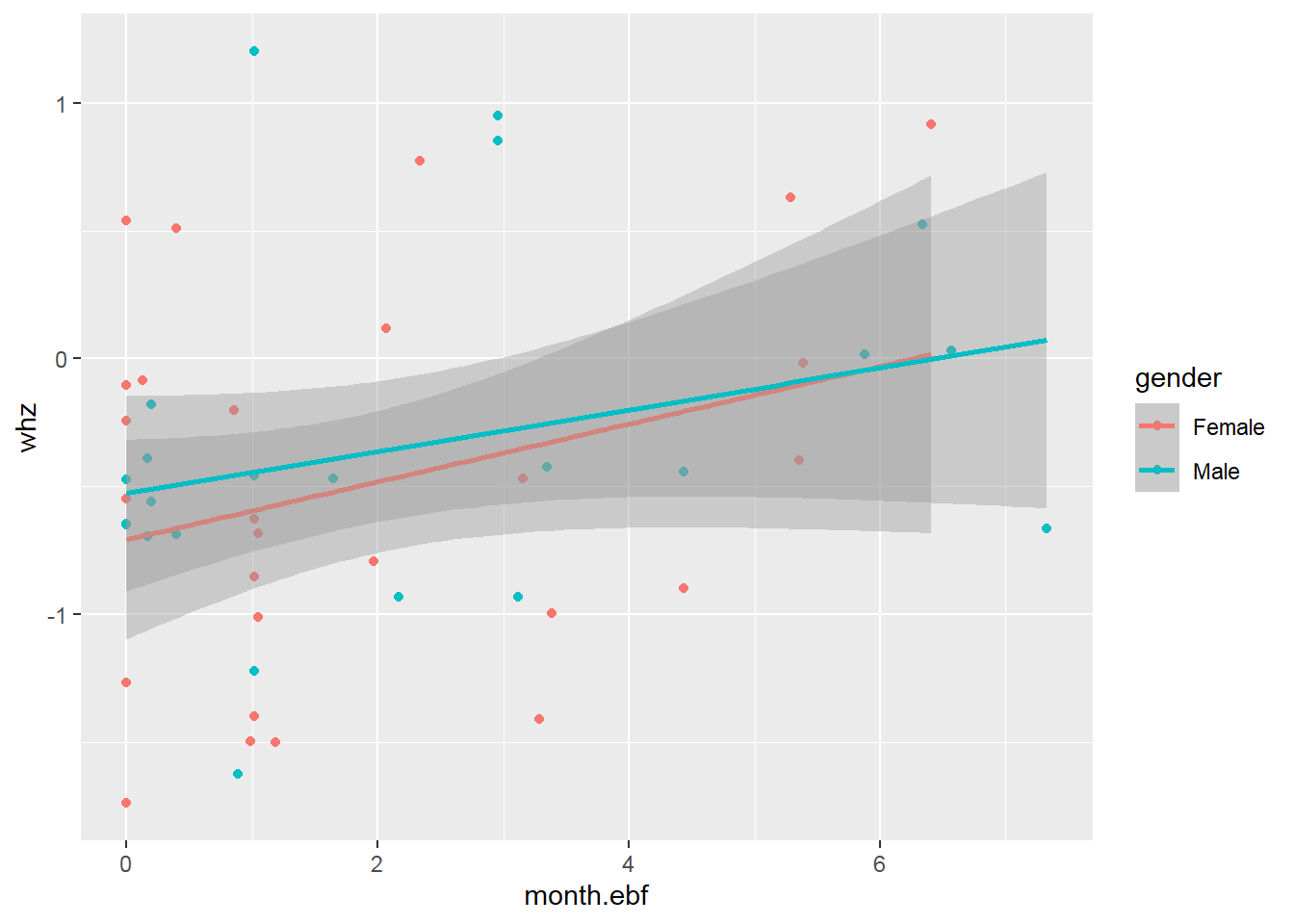
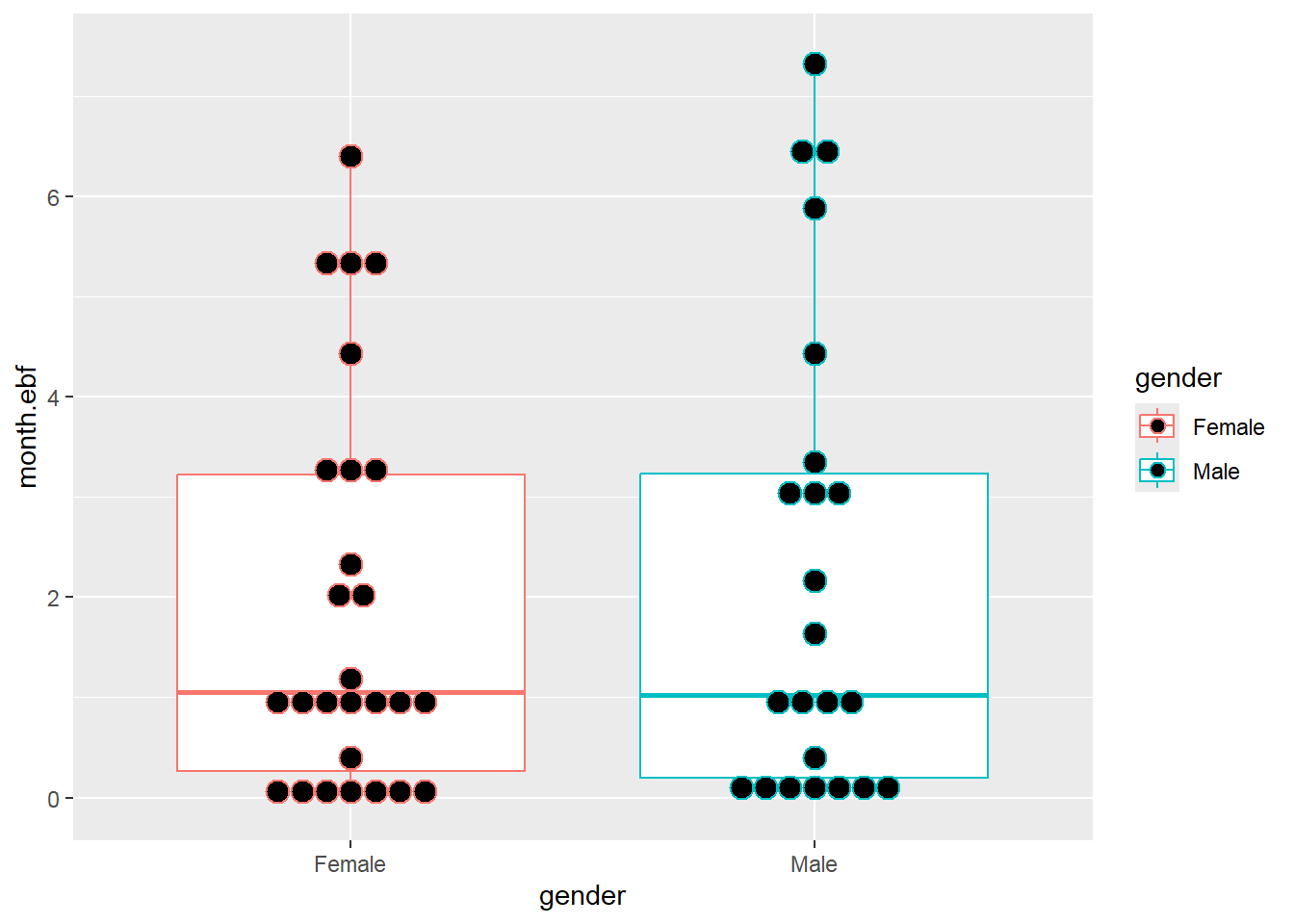
2.8.7 Pie Charts
dat2a %>%
group_by(gender) %>%
summarise(count=n()) %>%
ggplot(aes(x="",y=count,fill=gender)) +
geom_bar(stat = "identity") +
coord_polar("y", start = 0)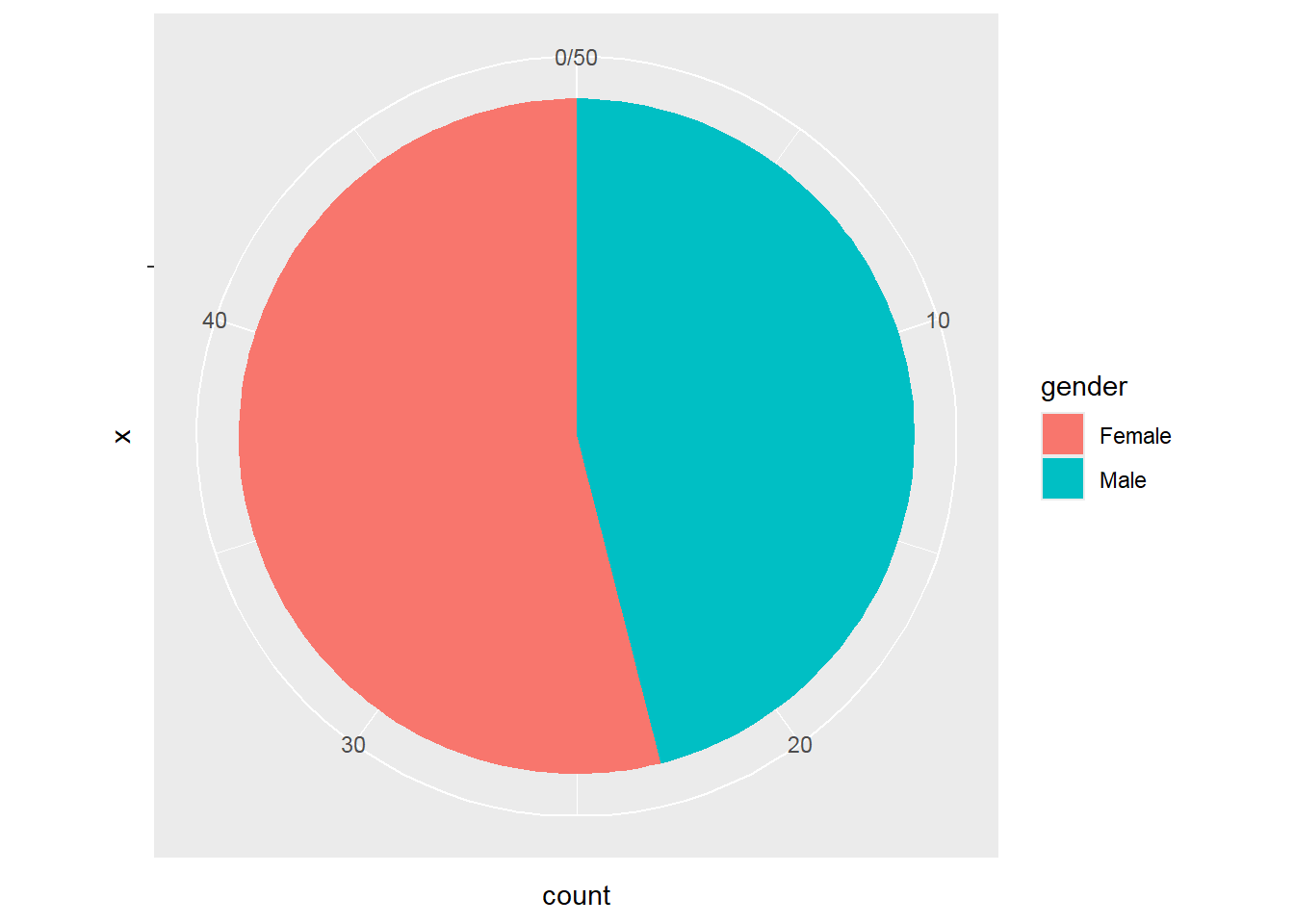
2.9 Summary statistics
2.9.1 Basic descriptive summary functions
# Summary of variable month.ebf
dat2a %>%
summarise(
count = n(),
mean = mean(month.ebf, na.rm = TRUE),
sd = sd(month.ebf, na.rm = TRUE),
median = median(month.ebf, na.rm = TRUE),
IQR = IQR(month.ebf, na.rm = TRUE))## count mean sd median IQR
## 1 50 2.071644 2.154823 1.034836 3.0552312.9.1.1 Summary by group
# Summary of month.ebf by cohort
dat2a %>% group_by(cohort) %>%
summarise(
count = n(),
mean = mean(month.ebf, na.rm = TRUE),
sd = sd(month.ebf, na.rm = TRUE),
median = median(month.ebf, na.rm = TRUE),
IQR = IQR(month.ebf, na.rm = TRUE))## # A tibble: 2 × 6
## cohort count mean sd median IQR
## <chr> <int> <dbl> <dbl> <dbl> <dbl>
## 1 Healthy Singleton Birth Cohort 25 3.45 2.24 3.29 3.71
## 2 Healthy Twins & Triplets 25 0.694 0.706 0.854 1.022.9.1.2 Summary by subgroup of another group
dat2a %>% filter(cohort == "Healthy Twins & Triplets") %>%
group_by(gender) %>%
summarise(
count = n(),
mean = mean(month.ebf, na.rm = TRUE),
sd = sd(month.ebf, na.rm = TRUE),
median = median(month.ebf, na.rm = TRUE),
IQR = IQR(month.ebf, na.rm = TRUE))## # A tibble: 2 × 6
## gender count mean sd median IQR
## <chr> <int> <dbl> <dbl> <dbl> <dbl>
## 1 Female 18 0.772 0.770 0.920 1.04
## 2 Male 7 0.493 0.498 0.197 0.9202.9.1.3 More filter
dat2a %>% filter(cohort == "Healthy Twins & Triplets") %>%
filter(!is.na(zygosity)) %>%
group_by(gender) %>%
summarise(
count = n(),
mean = mean(month.ebf, na.rm = TRUE),
sd = sd(month.ebf, na.rm = TRUE),
median = median(month.ebf, na.rm = TRUE),
IQR = IQR(month.ebf, na.rm = TRUE))## # A tibble: 2 × 6
## gender count mean sd median IQR
## <chr> <int> <dbl> <dbl> <dbl> <dbl>
## 1 Female 16 0.749 0.816 0.690 1.03
## 2 Male 7 0.493 0.498 0.197 0.9202.9.2 Basic statistical test examples
2.9.2.1 Continuous variables: parametric
## Min. 1st Qu. Median Mean 3rd Qu. Max.
## -1.73842 -0.88598 -0.47063 -0.42223 -0.03458 1.20133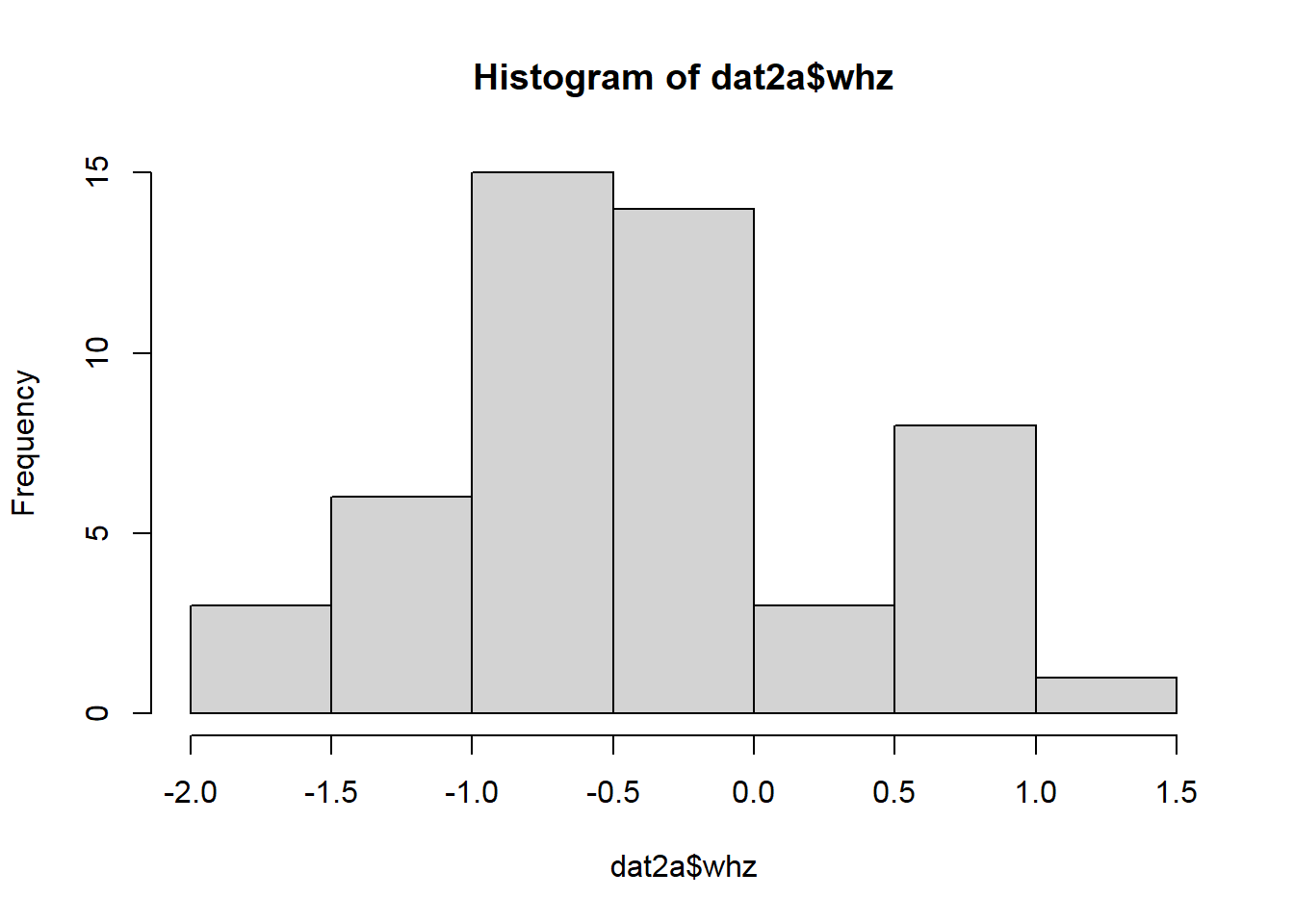
##
## Welch Two Sample t-test
##
## data: whz by gender
## t = -0.7285, df = 47.802, p-value = 0.4699
## alternative hypothesis: true difference in means between group Female and group Male is not equal to 0
## 95 percent confidence interval:
## -0.5572199 0.2608470
## sample estimates:
## mean in group Female mean in group Male
## -0.4903957 -0.34220922.9.2.2 Continuous variables: non-parametric
## Min. 1st Qu. Median Mean 3rd Qu. Max.
## 0.0000 0.1971 1.0348 2.0716 3.2523 7.3260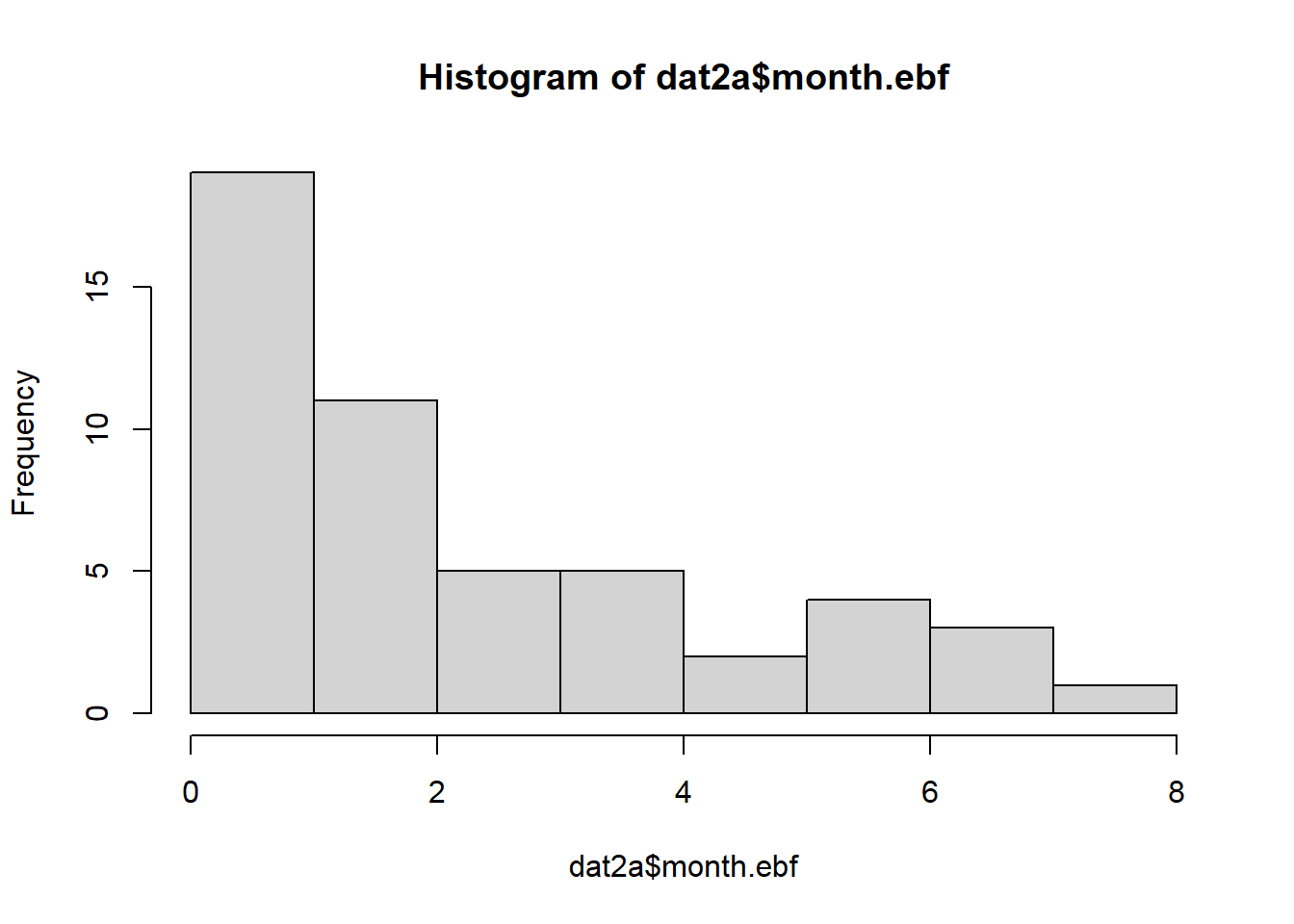
##
## Kruskal-Wallis rank sum test
##
## data: month.ebf by gender
## Kruskal-Wallis chi-squared = 0.12372, df = 1, p-value = 0.725##
## Wilcoxon rank sum test with continuity correction
##
## data: month.ebf by gender
## W = 292.5, p-value = 0.7324
## alternative hypothesis: true location shift is not equal to 02.9.2.3 Categorical variables
##
## Female Male
## Healthy Singleton Birth Cohort 9 16
## Healthy Twins & Triplets 18 7##
## Pearson's Chi-squared test with Yates' continuity correction
##
## data: dat2a$cohort and dat2a$gender
## X-squared = 5.153, df = 1, p-value = 0.02321##
## Fisher's Exact Test for Count Data
##
## data: dat2a$cohort and dat2a$gender
## p-value = 0.02224
## alternative hypothesis: true odds ratio is not equal to 1
## 95 percent confidence interval:
## 0.05544835 0.83324786
## sample estimates:
## odds ratio
## 0.22616442.9.3 Summary statistic table
2.9.3.1 Packages for quick, nice summary table
The “arsenal” package provides handy R functions for large-scale statistical summaries (Heinzen et al. 2021).
#install.packages(c("Deducer","arsenal","qwraps2","table1","tangram","purrrlyr","Gmisc"))
library(arsenal)
ca.vars<-c("cohort","gender","zygosity")
co.vars<-c("whz", "waz", "haz","age.first.fe","age.last.fe","no.fe.sam","sam.int.mean",
"month.ebf","age.first.solid","no.diarhea.yr","pc.days.diarrhea","frac.sam.atb.7day")
varuse<-c(co.vars,ca.vars[!ca.vars %in% "cohort"])
mylabels <-as.list(varuse)
names(mylabels)<-varuse
tab1 <- tableby(as.formula(paste("cohort",paste(varuse,collapse="+"),sep="~")),
data = dat2a)
kable(summary(tab1,
labelTranslations = mylabels,
text=TRUE))| Healthy Singleton Birth Cohort (N=25) | Healthy Twins & Triplets (N=25) | Total (N=50) | p value | |
|---|---|---|---|---|
| whz | 0.683 | |||
| - Mean (SD) | -0.380 (0.727) | -0.464 (0.724) | -0.422 (0.719) | |
| - Range | -1.623 - 0.949 | -1.738 - 1.201 | -1.738 - 1.201 | |
| waz | < 0.001 | |||
| - Mean (SD) | -0.483 (0.806) | -2.546 (0.561) | -1.515 (1.248) | |
| - Range | -1.741 - 1.670 | -3.755 - -1.392 | -3.755 - 1.670 | |
| haz | < 0.001 | |||
| - Mean (SD) | -0.483 (0.806) | -3.231 (0.956) | -1.857 (1.641) | |
| - Range | -1.741 - 1.670 | -4.659 - -1.412 | -4.659 - 1.670 | |
| age.first.fe | 0.008 | |||
| - Mean (SD) | 6.360 (2.782) | 13.200 (12.124) | 9.780 (9.366) | |
| - Range | 3.000 - 15.000 | 1.000 - 37.000 | 1.000 - 37.000 | |
| age.last.fe | < 0.001 | |||
| - Mean (SD) | 710.400 (12.430) | 490.880 (152.553) | 600.640 (154.167) | |
| - Range | 700.000 - 738.000 | 286.000 - 730.000 | 286.000 - 738.000 | |
| no.fe.sam | 0.001 | |||
| - Mean (SD) | 21.920 (2.019) | 17.920 (5.338) | 19.920 (4.476) | |
| - Range | 18.000 - 24.000 | 9.000 - 26.000 | 9.000 - 26.000 | |
| sam.int.mean | < 0.001 | |||
| - Mean (SD) | 34.003 (3.813) | 28.677 (3.090) | 31.340 (4.363) | |
| - Range | 30.087 - 42.294 | 22.562 - 35.000 | 22.562 - 42.294 | |
| month.ebf | < 0.001 | |||
| - Mean (SD) | 3.449 (2.242) | 0.694 (0.706) | 2.072 (2.155) | |
| - Range | 0.000 - 7.326 | 0.000 - 2.332 | 0.000 - 7.326 | |
| age.first.solid | 0.254 | |||
| - Mean (SD) | 6.352 (2.236) | 7.028 (1.884) | 6.690 (2.074) | |
| - Range | 1.216 - 9.790 | 3.088 - 9.396 | 1.216 - 9.790 | |
| no.diarhea.yr | < 0.001 | |||
| - Mean (SD) | 4.278 (2.135) | 1.664 (1.345) | 2.971 (2.205) | |
| - Range | 0.520 - 8.814 | 0.000 - 4.986 | 0.000 - 8.814 | |
| pc.days.diarrhea | 0.007 | |||
| - Mean (SD) | 4.090 (2.223) | 2.159 (2.601) | 3.125 (2.585) | |
| - Range | 0.142 - 10.826 | 0.000 - 9.563 | 0.000 - 10.826 | |
| frac.sam.atb.7day | 0.070 | |||
| - Mean (SD) | 0.219 (0.145) | 0.148 (0.125) | 0.183 (0.139) | |
| - Range | 0.000 - 0.500 | 0.000 - 0.462 | 0.000 - 0.500 | |
| gender | 0.011 | |||
| - Female | 9 (36.0%) | 18 (72.0%) | 27 (54.0%) | |
| - Male | 16 (64.0%) | 7 (28.0%) | 23 (46.0%) | |
| zygosity | < 0.001 | |||
| - N-Miss | 0 | 2 | 2 | |
| - DZ | 0 (0.0%) | 12 (52.2%) | 12 (25.0%) | |
| - Fraternal co-twin in set of triplets | 0 (0.0%) | 1 (4.3%) | 1 (2.1%) | |
| - MZ | 0 (0.0%) | 8 (34.8%) | 8 (16.7%) | |
| - MZ co-twin in set of triplets | 0 (0.0%) | 2 (8.7%) | 2 (4.2%) | |
| - singleton | 25 (100.0%) | 0 (0.0%) | 25 (52.1%) |
2.9.3.2 More comprehensive table (control)
my_controls <- tableby.control(
test = T,
total = T,
numeric.test = "kwt", cat.test = "fe",
numeric.stats = c("meansd", "medianq1q3", "range", "Nmiss2"),
cat.stats = c("countpct", "Nmiss2"),
stats.labels = list(
meansd = "Mean (SD)",
medianq1q3 = "Median (Q1, Q3)",
range = "Min - Max",
Nmiss2 = "Missing"
)
)
tab2 <- tableby(as.formula(paste("cohort",paste(varuse,collapse="+"),sep="~")),data = dat2a,
control = my_controls)
kable(summary(tab2,labelTranslations = mylabels, text=TRUE))| Healthy Singleton Birth Cohort (N=25) | Healthy Twins & Triplets (N=25) | Total (N=50) | p value | |
|---|---|---|---|---|
| whz | 0.705 | |||
| - Mean (SD) | -0.380 (0.727) | -0.464 (0.724) | -0.422 (0.719) | |
| - Median (Q1, Q3) | -0.468 (-0.897, 0.015) | -0.560 (-0.853, -0.104) | -0.471 (-0.886, -0.035) | |
| - Min - Max | -1.623 - 0.949 | -1.738 - 1.201 | -1.738 - 1.201 | |
| - Missing | 0 | 0 | 0 | |
| waz | < 0.001 | |||
| - Mean (SD) | -0.483 (0.806) | -2.546 (0.561) | -1.515 (1.248) | |
| - Median (Q1, Q3) | -0.706 (-1.031, -0.030) | -2.606 (-2.879, -2.295) | -1.512 (-2.604, -0.719) | |
| - Min - Max | -1.741 - 1.670 | -3.755 - -1.392 | -3.755 - 1.670 | |
| - Missing | 0 | 0 | 0 | |
| haz | < 0.001 | |||
| - Mean (SD) | -0.483 (0.806) | -3.231 (0.956) | -1.857 (1.641) | |
| - Median (Q1, Q3) | -0.706 (-1.031, -0.030) | -3.437 (-4.032, -2.551) | -1.575 (-3.431, -0.719) | |
| - Min - Max | -1.741 - 1.670 | -4.659 - -1.412 | -4.659 - 1.670 | |
| - Missing | 0 | 0 | 0 | |
| age.first.fe | 0.077 | |||
| - Mean (SD) | 6.360 (2.782) | 13.200 (12.124) | 9.780 (9.366) | |
| - Median (Q1, Q3) | 5.000 (5.000, 8.000) | 8.000 (5.000, 15.000) | 6.000 (5.000, 9.750) | |
| - Min - Max | 3.000 - 15.000 | 1.000 - 37.000 | 1.000 - 37.000 | |
| - Missing | 0 | 0 | 0 | |
| age.last.fe | < 0.001 | |||
| - Mean (SD) | 710.400 (12.430) | 490.880 (152.553) | 600.640 (154.167) | |
| - Median (Q1, Q3) | 704.000 (701.000, 715.000) | 455.000 (366.000, 638.000) | 700.500 (455.000, 706.000) | |
| - Min - Max | 700.000 - 738.000 | 286.000 - 730.000 | 286.000 - 738.000 | |
| - Missing | 0 | 0 | 0 | |
| no.fe.sam | 0.011 | |||
| - Mean (SD) | 21.920 (2.019) | 17.920 (5.338) | 19.920 (4.476) | |
| - Median (Q1, Q3) | 23.000 (21.000, 23.000) | 18.000 (13.000, 22.000) | 21.000 (18.000, 23.000) | |
| - Min - Max | 18.000 - 24.000 | 9.000 - 26.000 | 9.000 - 26.000 | |
| - Missing | 0 | 0 | 0 | |
| sam.int.mean | < 0.001 | |||
| - Mean (SD) | 34.003 (3.813) | 28.677 (3.090) | 31.340 (4.363) | |
| - Median (Q1, Q3) | 33.143 (31.409, 35.350) | 28.154 (27.048, 30.350) | 30.477 (28.290, 33.271) | |
| - Min - Max | 30.087 - 42.294 | 22.562 - 35.000 | 22.562 - 42.294 | |
| - Missing | 0 | 0 | 0 | |
| month.ebf | < 0.001 | |||
| - Mean (SD) | 3.449 (2.242) | 0.694 (0.706) | 2.072 (2.155) | |
| - Median (Q1, Q3) | 3.285 (1.643, 5.355) | 0.854 (0.000, 1.018) | 1.035 (0.197, 3.252) | |
| - Min - Max | 0.000 - 7.326 | 0.000 - 2.332 | 0.000 - 7.326 | |
| - Missing | 0 | 0 | 0 | |
| age.first.solid | 0.388 | |||
| - Mean (SD) | 6.352 (2.236) | 7.028 (1.884) | 6.690 (2.074) | |
| - Median (Q1, Q3) | 6.209 (4.599, 8.213) | 7.063 (6.078, 8.476) | 6.932 (4.920, 8.361) | |
| - Min - Max | 1.216 - 9.790 | 3.088 - 9.396 | 1.216 - 9.790 | |
| - Missing | 0 | 0 | 0 | |
| no.diarhea.yr | < 0.001 | |||
| - Mean (SD) | 4.278 (2.135) | 1.664 (1.345) | 2.971 (2.205) | |
| - Median (Q1, Q3) | 3.957 (3.008, 5.170) | 1.276 (0.802, 2.003) | 2.916 (1.271, 4.054) | |
| - Min - Max | 0.520 - 8.814 | 0.000 - 4.986 | 0.000 - 8.814 | |
| - Missing | 0 | 0 | 0 | |
| pc.days.diarrhea | < 0.001 | |||
| - Mean (SD) | 4.090 (2.223) | 2.159 (2.601) | 3.125 (2.585) | |
| - Median (Q1, Q3) | 3.930 (2.996, 4.755) | 1.359 (0.440, 2.740) | 2.742 (1.163, 4.314) | |
| - Min - Max | 0.142 - 10.826 | 0.000 - 9.563 | 0.000 - 10.826 | |
| - Missing | 0 | 0 | 0 | |
| frac.sam.atb.7day | 0.109 | |||
| - Mean (SD) | 0.219 (0.145) | 0.148 (0.125) | 0.183 (0.139) | |
| - Median (Q1, Q3) | 0.217 (0.087, 0.318) | 0.111 (0.077, 0.167) | 0.119 (0.081, 0.284) | |
| - Min - Max | 0.000 - 0.500 | 0.000 - 0.462 | 0.000 - 0.500 | |
| - Missing | 0 | 0 | 0 | |
| gender | 0.022 | |||
| - Female | 9 (36.0%) | 18 (72.0%) | 27 (54.0%) | |
| - Male | 16 (64.0%) | 7 (28.0%) | 23 (46.0%) | |
| - Missing | 0 | 0 | 0 | |
| zygosity | < 0.001 | |||
| - DZ | 0 (0.0%) | 12 (52.2%) | 12 (25.0%) | |
| - Fraternal co-twin in set of triplets | 0 (0.0%) | 1 (4.3%) | 1 (2.1%) | |
| - MZ | 0 (0.0%) | 8 (34.8%) | 8 (16.7%) | |
| - MZ co-twin in set of triplets | 0 (0.0%) | 2 (8.7%) | 2 (4.2%) | |
| - singleton | 25 (100.0%) | 0 (0.0%) | 25 (52.1%) | |
| - Missing | 0 | 2 | 2 |
2.10 Practice
Write R code to:
- Import sheet 2 of excel data file “nature13421-s2.xlsx”
- Rename column names to sensible names, remove redundant rows
- Save your cleaned data to local disc and load saved data for use
- Make a table showing number of fecal sample per each child ID
- Summarize number of fecal samples in all child ID and by cohort of singletons vs twins/triplets
- Plot number of fecal samples by cohort of singletons vs twins/triplets
2.11 Practice review
2.11.1 The requirements
Import sheet 2 of excel data file (Chan et al. 2023):
library(rio)
#import all sheets
list1<-import_list(paste(fdir1,"nature13421-s2.xlsx",sep=""))
#get sheet 2
sh2<-list1[[2]]
#import sheet 2 only
sh2<-import(paste(fdir1,"nature13421-s2.xlsx",sep=""),sheet=2,skip=1) Rename column names to sensible names, remove redundant rows:
library(tidyverse)
#colnames(sh2)
colnames(sh2)[!is.na(sh2[1,])]<-sh2[1,][!is.na(sh2[1,])]
#or just do all at a time
oldname<-colnames(sh2)
newname<-c("cohort",
"family.id",
"child.id",
"sample.id",
"age.d",
"age.m",
"whz","waz","haz",
"breast.milk",
"formula",
"solid",
"diarrhea",
"antibiotic.7d",
"medication",
"no.sequence",
"serun.id",
"barcode")
sh2<-sh2 %>% rename_at(vars(oldname), ~newname)
#colnames(sh2)
#remove unused rows
sh2<-sh2 %>%
filter(!is.na(family.id))You can save your cleaned data to local disc and load saved data for use:
#save data
#write.csv(sh2,file=paste(fdir1,"sh2.csv",sep=""),row.names=F)
#sh2<-read.csv(paste(fdir1,"sh2.csv",sep=""))Make a table showing number of fecal sample per each child ID and summarize number of fecal samples in all child ID:
nsam<-sh2 %>%
group_by(child.id) %>%
summarise(no.sam=n())
#View(nsam)
nsam %>%
summarise(
mean = mean(no.sam, na.rm = TRUE),
sd = sd(no.sam, na.rm = TRUE),
median = median(no.sam, na.rm = TRUE),
IQR = IQR(no.sam, na.rm = TRUE))## # A tibble: 1 × 4
## mean sd median IQR
## <dbl> <dbl> <dbl> <dbl>
## 1 19.9 4.48 21 5Summarize number of fecal samples per childid by cohort of singletons vs twins/triplets:
nsam<-sh2 %>%
group_by(cohort,child.id) %>%
summarise(no.sam=n()) %>%
summarise(
mean = mean(no.sam, na.rm = TRUE),
sd = sd(no.sam, na.rm = TRUE),
median = median(no.sam, na.rm = TRUE),
IQR = IQR(no.sam, na.rm = TRUE))
nsam ## # A tibble: 2 × 5
## cohort mean sd median IQR
## <chr> <dbl> <dbl> <int> <dbl>
## 1 Healthy Singleton Birth Cohort 21.9 2.02 23 2
## 2 Healthy Twins & Triplets 17.9 5.34 18 9Boxplot number of fecal samples by cohort of singletons vs twins/triplets:
sh2 %>%
group_by(cohort,child.id) %>%
summarise(no.sam=n()) %>%
ggplot(aes(x=cohort,y=no.sam,fill=cohort)) +
geom_boxplot()+
ylab("Number of fecal samples")+
geom_dotplot(binaxis='y', stackdir='center', dotsize=1)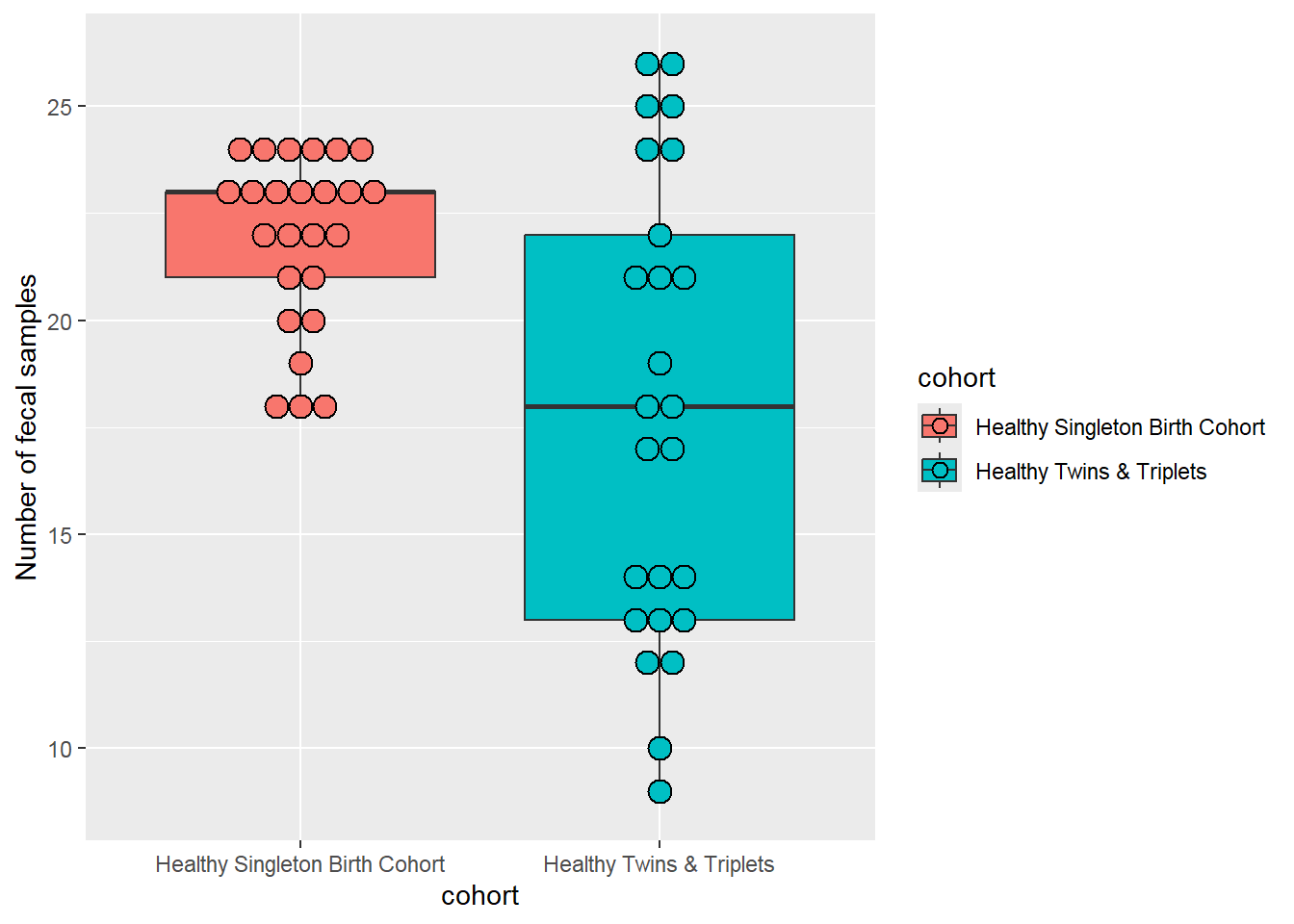
Violin Plot: similar to box plot but shows the density within groups, not much info provided as in box plot:
sh2 %>%
group_by(cohort,child.id) %>%
summarise(no.sam=n()) %>%
ggplot(aes(x=cohort,y=no.sam,fill=cohort)) +
geom_violin()+
ylab("Number of fecal samples")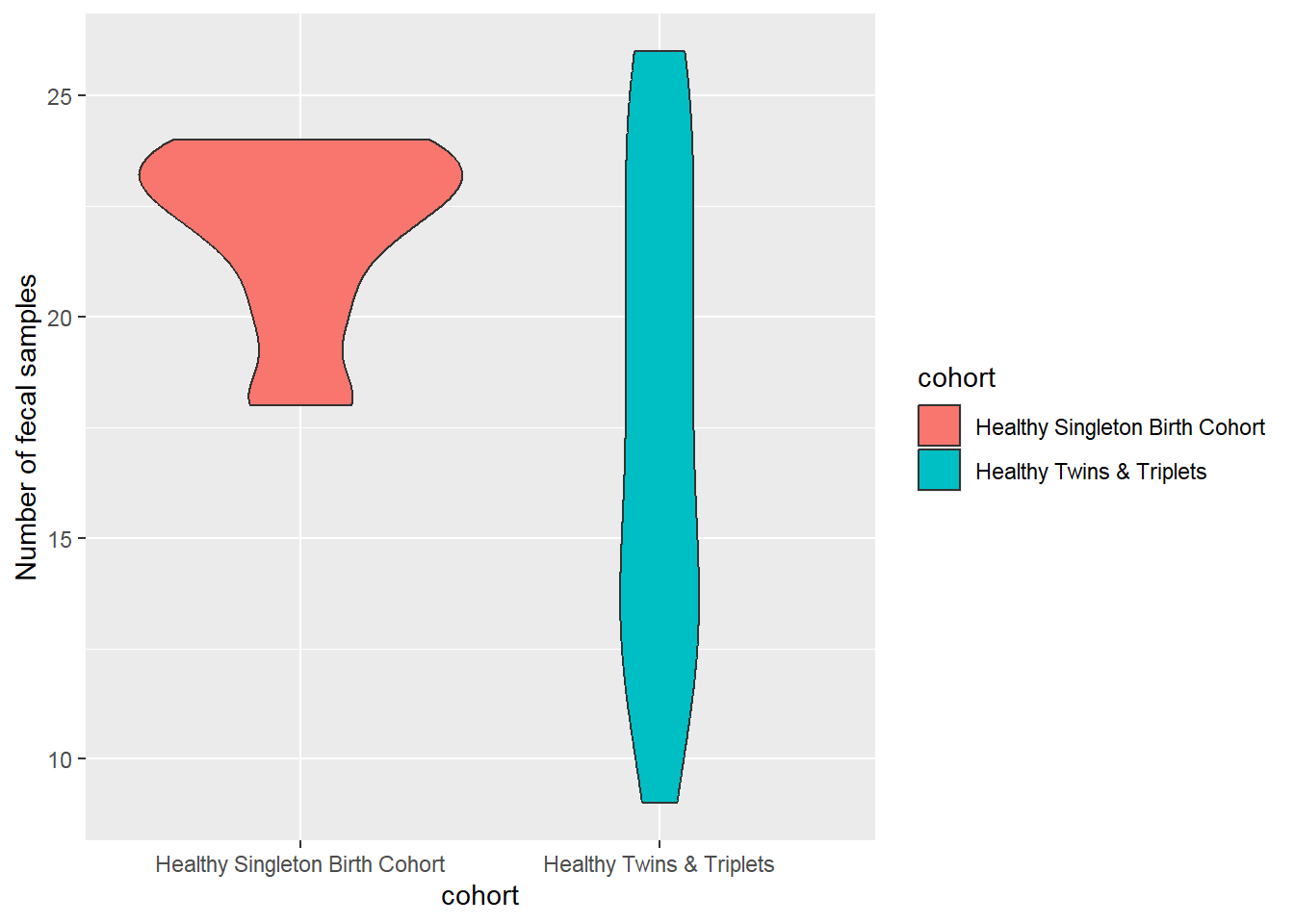
Bar plot number of fecal samples by cohort of singletons vs twins/triplets and summary of number of fecal samples:
nsam<-sh2 %>%
group_by(cohort,child.id) %>%
summarise(no.sam=n()) %>%
#group_by(cohort) %>%
summarise(
no.child = n(),
mean = mean(no.sam, na.rm = TRUE),
sd = sd(no.sam, na.rm = TRUE),
se=sd/sqrt(n()),
ci=1.96*se,
median = median(no.sam, na.rm = TRUE),
IQR = IQR(no.sam, na.rm = TRUE))
nsam## # A tibble: 2 × 8
## cohort no.child mean sd se ci median IQR
## <chr> <int> <dbl> <dbl> <dbl> <dbl> <int> <dbl>
## 1 Healthy Singleton Birth Cohort 25 21.9 2.02 0.404 0.791 23 2
## 2 Healthy Twins & Triplets 25 17.9 5.34 1.07 2.09 18 9Bar plot with error bar:
ggplot(nsam, aes(x=cohort, y=mean, fill=cohort)) +
geom_bar(position=position_dodge(), stat="identity") +
geom_errorbar(aes(ymin=mean-se, ymax=mean+se),
width=.2, # Width of the error bars
position=position_dodge(.9))+
ylab("Number of fecal samples (mean,se)")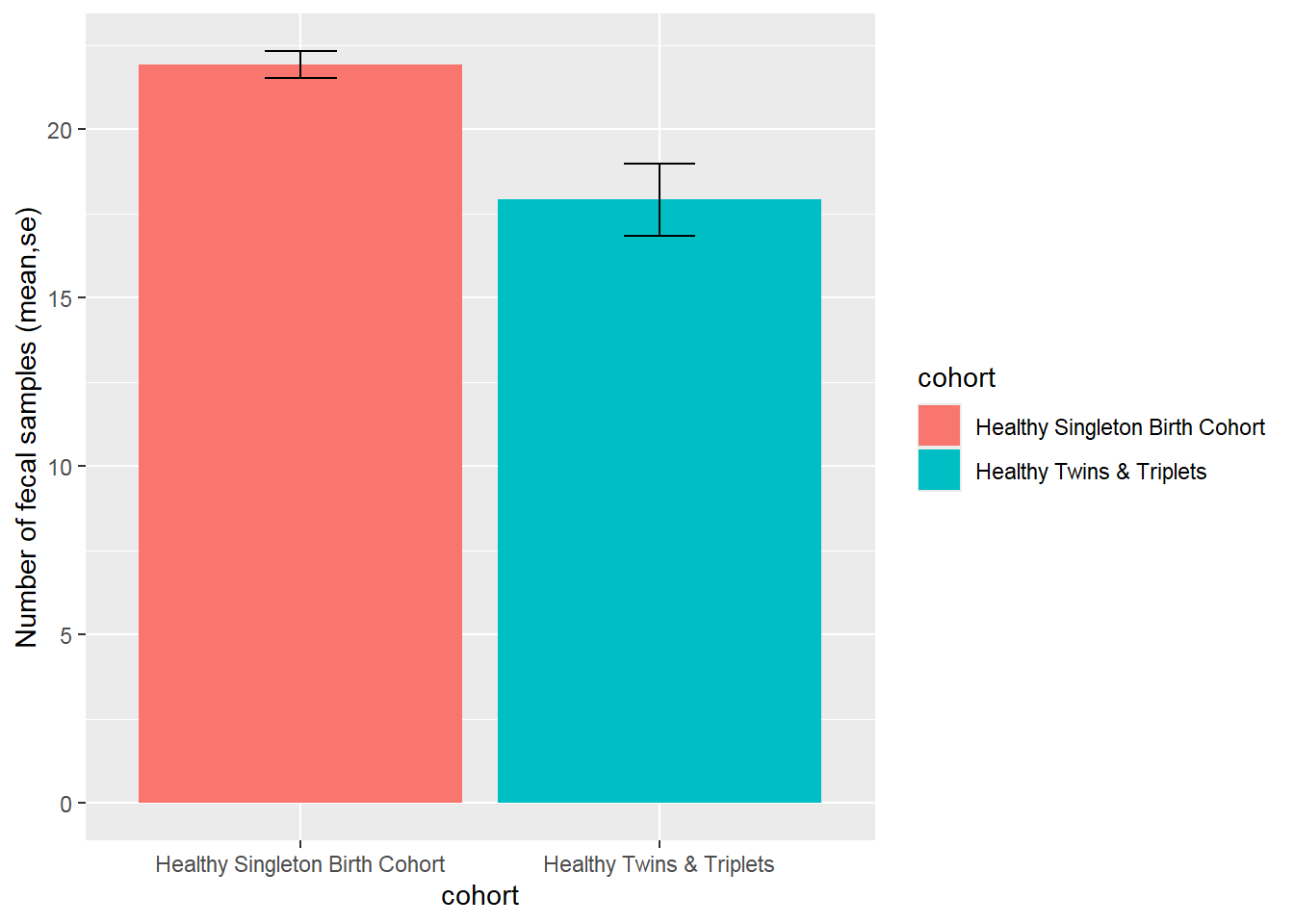
Bar plot with 95%CI:
ggplot(nsam, aes(x=cohort, y=mean, fill=cohort)) +
geom_bar(position=position_dodge(), stat="identity") +
geom_errorbar(aes(ymin=mean-ci, ymax=mean+ci),
width=.2, # Width of the error bars
position=position_dodge(.9))+
ylab("Number of fecal samples (mean, 95%CI)")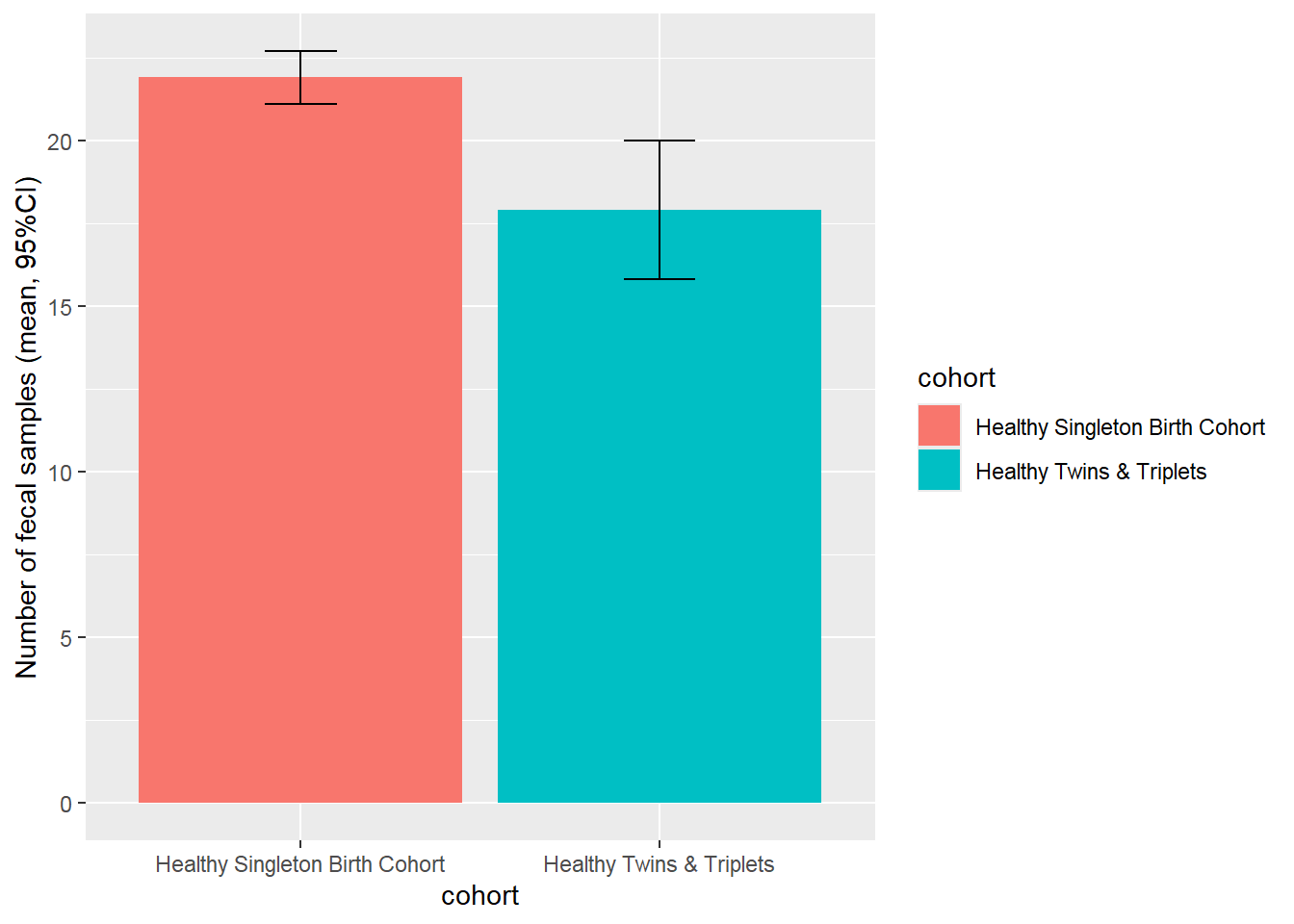
Histogram number of fecal samples by cohort of singletons vs twins/triplets:
sh2 %>%
group_by(cohort,child.id) %>%
summarise(no.sam=n()) %>%
ggplot(aes(no.sam,y=..density..,fill=cohort))+
geom_histogram(bins=10,alpha=0.5, position="identity")+
geom_density(aes(no.sam,color=cohort),alpha=0.5)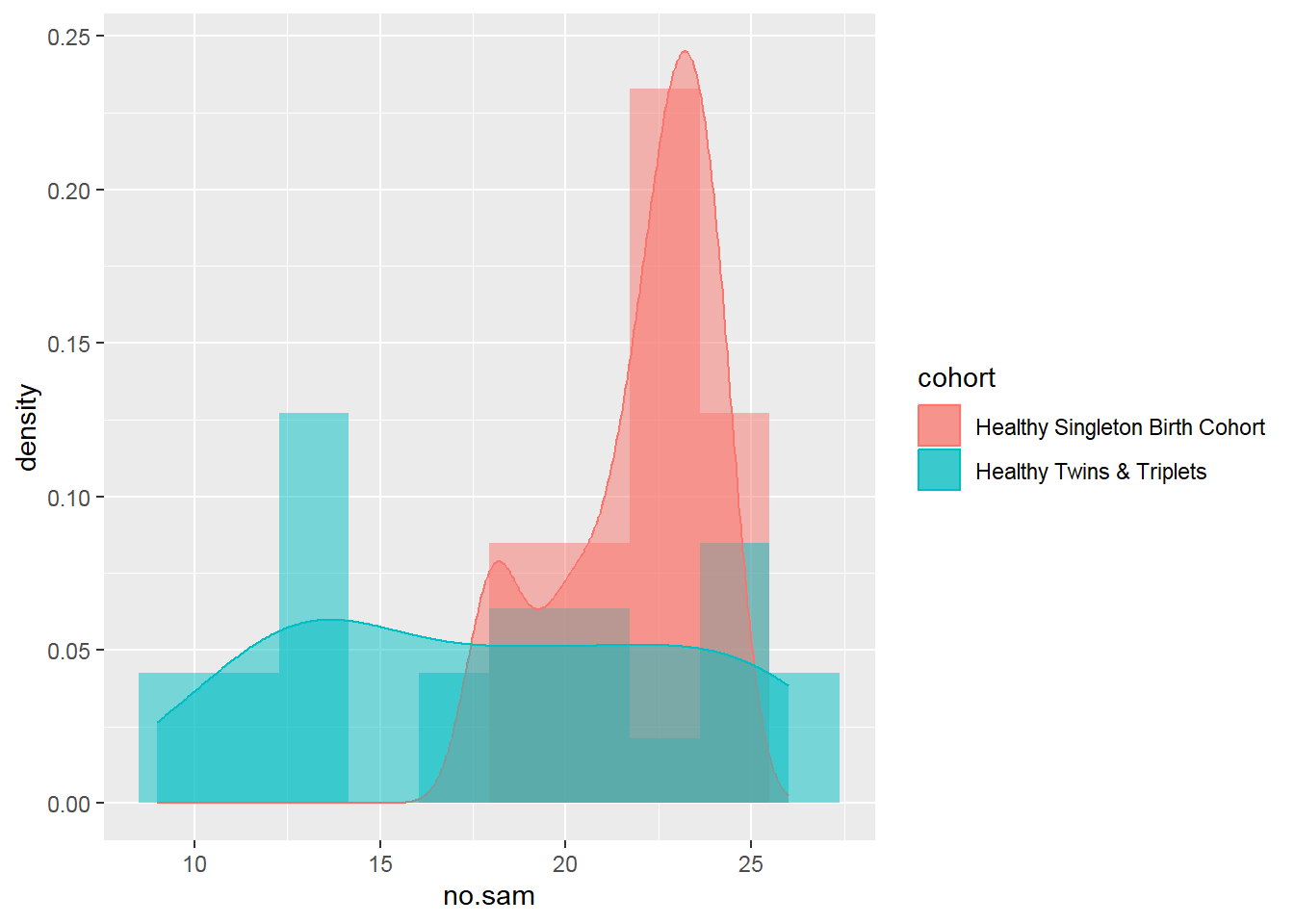
2.11.2 Further exploration
Basic statistical test:
## # A tibble: 6 × 3
## # Groups: cohort [1]
## cohort child.id no.sam
## <chr> <chr> <int>
## 1 Healthy Singleton Birth Cohort Bgsng7018 21
## 2 Healthy Singleton Birth Cohort Bgsng7035 23
## 3 Healthy Singleton Birth Cohort Bgsng7052 20
## 4 Healthy Singleton Birth Cohort Bgsng7063 20
## 5 Healthy Singleton Birth Cohort Bgsng7071 18
## 6 Healthy Singleton Birth Cohort Bgsng7082 18##
## Wilcoxon rank sum test with continuity correction
##
## data: no.sam by cohort
## W = 443.5, p-value = 0.01092
## alternative hypothesis: true location shift is not equal to 0##
## Kruskal-Wallis rank sum test
##
## data: no.sam by cohort
## Kruskal-Wallis chi-squared = 6.5277, df = 1, p-value = 0.01062##
## Welch Two Sample t-test
##
## data: no.sam by cohort
## t = 3.5045, df = 30.73, p-value = 0.001427
## alternative hypothesis: true difference in means between group Healthy Singleton Birth Cohort and group Healthy Twins & Triplets is not equal to 0
## 95 percent confidence interval:
## 1.671264 6.328736
## sample estimates:
## mean in group Healthy Singleton Birth Cohort mean in group Healthy Twins & Triplets
## 21.92 17.92Which test is appropriate? => Please see Chapter “Basic Statistics” of this book.
Statistical test for difference in data type between sheet 1 and sheet 2?
- Sheet 1 is cross sectional data.
- Sheet 2 is longitudinal data: many observations per childid. Therefore, different ways of data description and analysis may be used.
Longitudinal data visualization with spaghetti plot:
sh2 %>%
filter(!is.na(whz)) %>%
ggplot()+
geom_point(aes(x = age.m, y = whz, group = child.id, colour=cohort))+
geom_line(aes(x = age.m, y = whz, group = child.id, colour=cohort),size=0.3)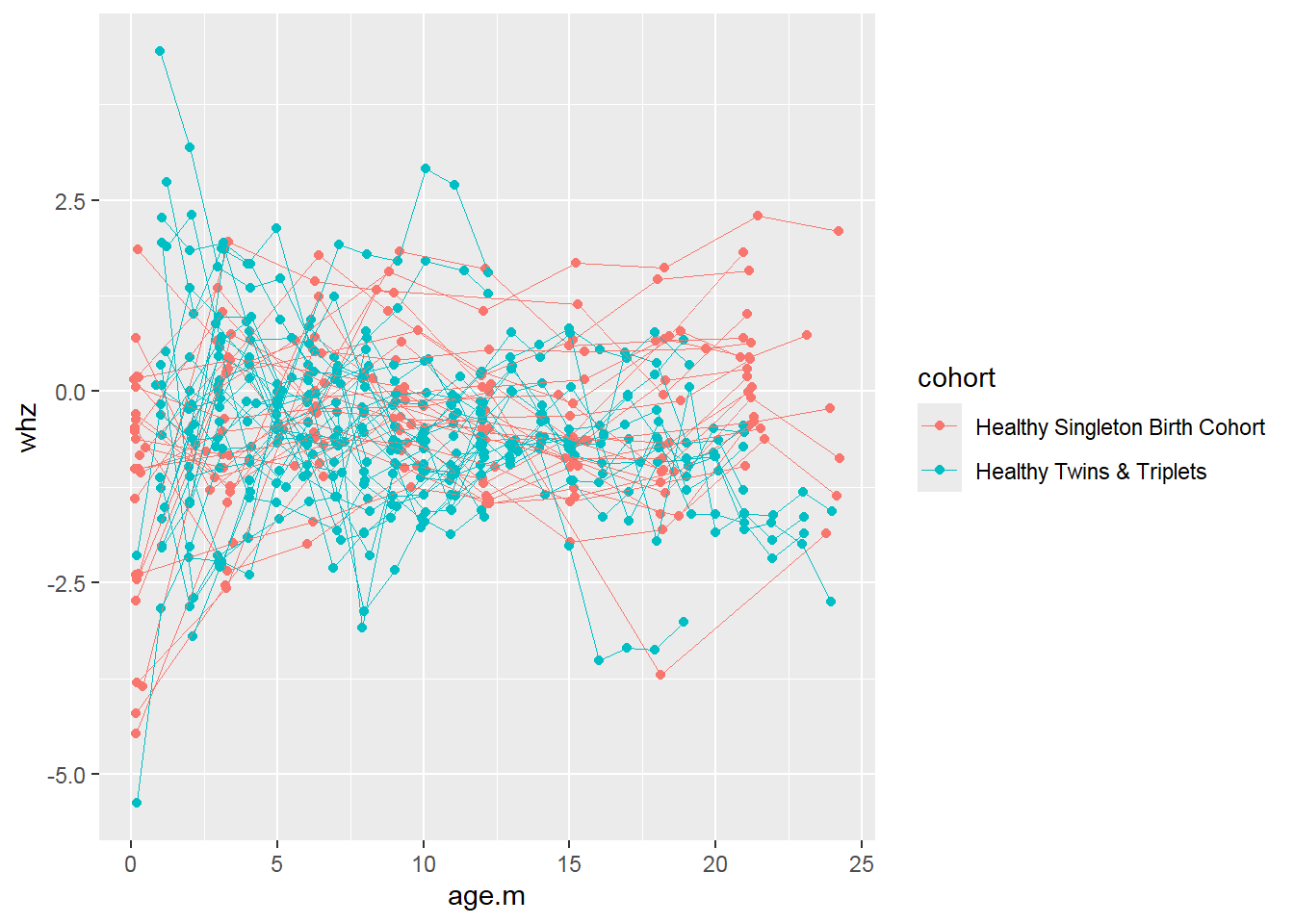
More exploration with waz:
sh2 %>%
filter(!is.na(waz)) %>%
ggplot()+
geom_point(aes(x = age.m, y = waz, group = child.id, colour=cohort))+
geom_line(aes(x = age.m, y = waz, group = child.id, colour=cohort),size=0.3)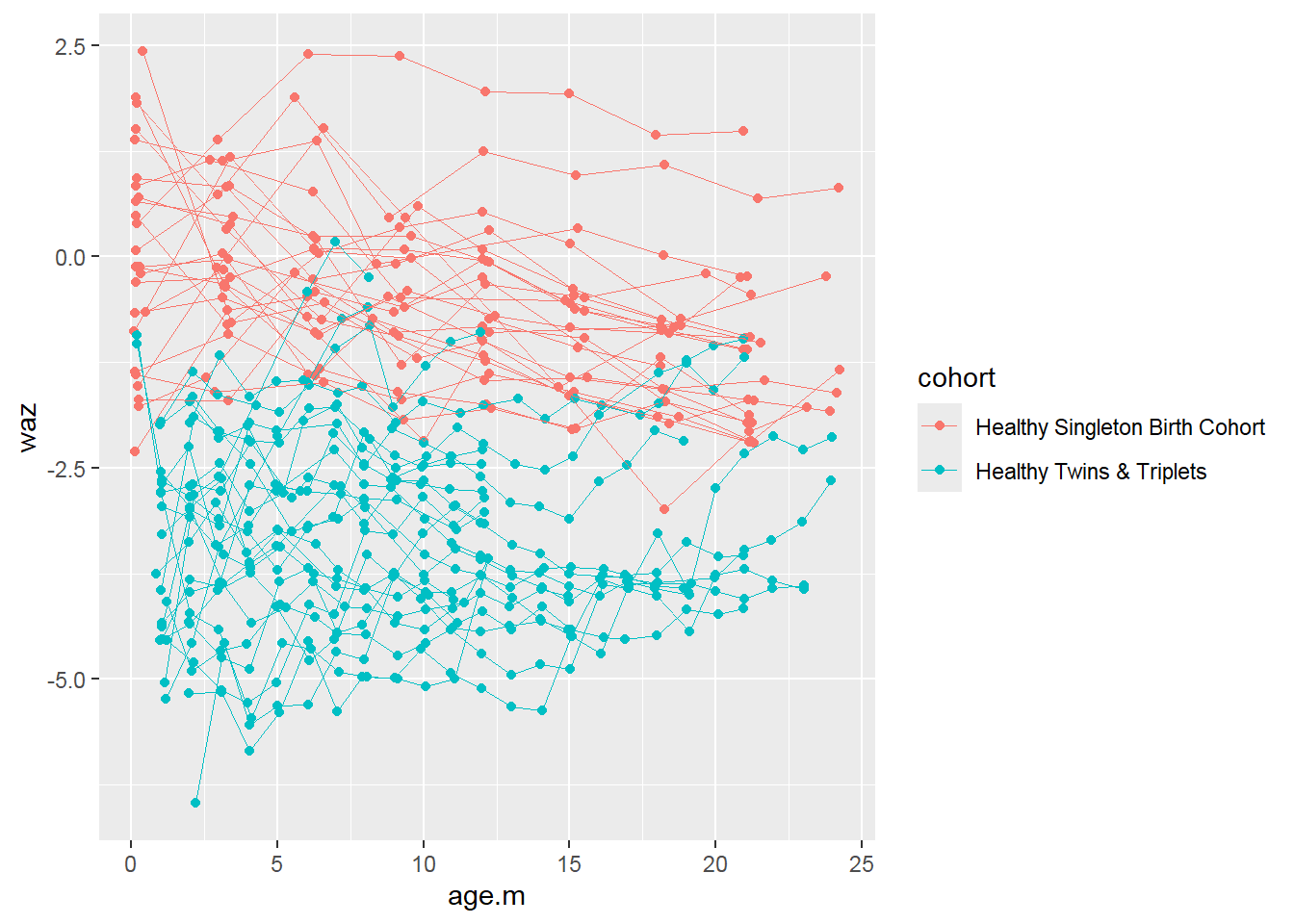
We can add fitted lines to the plot:
sh2 %>%
filter(!is.na(waz)) %>%
ggplot()+
geom_point(aes(x = age.m, y = waz, group = child.id, colour=cohort))+
geom_line(aes(x = age.m, y = waz, group = child.id, colour=cohort),size=0.3)+
stat_smooth(aes(x = age.m, y = waz, colour=cohort), method = 'glm',size=2)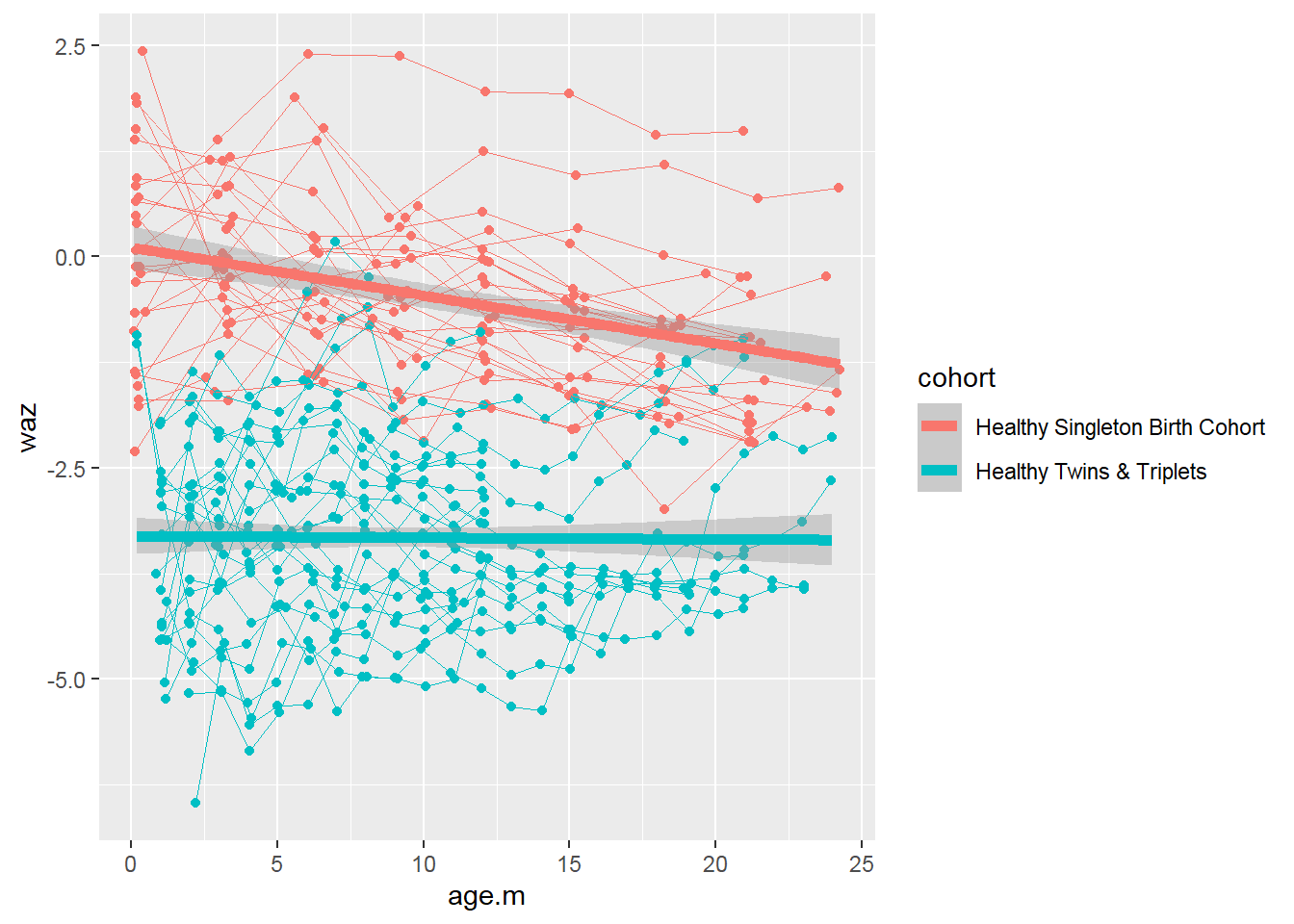
More exploration with haz:
sh2 %>%
filter(!is.na(haz)) %>%
ggplot()+
geom_point(aes(x = age.m, y = haz, group = child.id, colour=cohort))+
geom_line(aes(x = age.m, y = haz, group = child.id, colour=cohort),size=0.3)+
stat_smooth(aes(x = age.m, y = haz, colour=cohort), method = 'glm',size=2)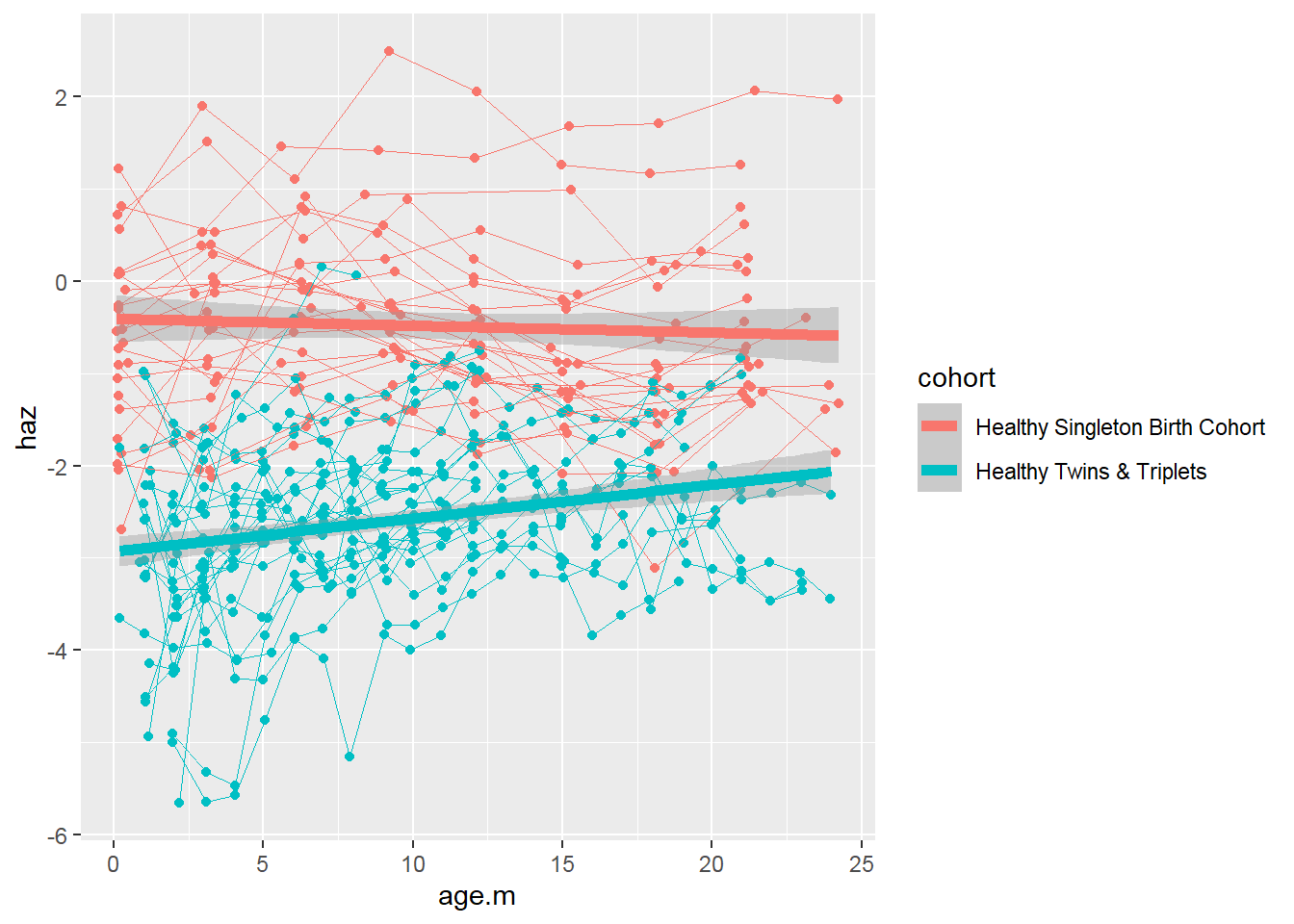
To test for the difference between two groups, check out the chapter “Longitudinal data analysis with R” in this book.
All-in-one spaghetti plot of three indices by cohort:
sh2 %>%
filter(!is.na(haz) & !is.na(waz) & !is.na(whz)) %>%
ggplot()+
#add each item to the plot
geom_point(aes(x = age.m, y = haz, group = child.id,color="haz"))+
geom_line(aes(x = age.m, y = haz, group = child.id,color="haz"),size=0.3)+
stat_smooth(aes(x = age.m, y = haz,color="haz"), method = 'glm',size=2)+
geom_point(aes(x = age.m, y = waz, group = child.id,color="waz"))+
geom_line(aes(x = age.m, y = waz, group = child.id,color="waz"),size=0.3)+
stat_smooth(aes(x = age.m, y = waz,color="waz"), method = 'glm',size=2)+
geom_point(aes(x = age.m, y = whz, group = child.id,color="whz"))+
geom_line(aes(x = age.m, y = whz, group = child.id,color="whz"),size=0.3)+
stat_smooth(aes(x = age.m, y = whz,color="whz"), method = 'glm',size=2)+
#divide the plot into multiple panels (cohort groups)
facet_grid(.~ cohort)+
theme(legend.title = element_blank())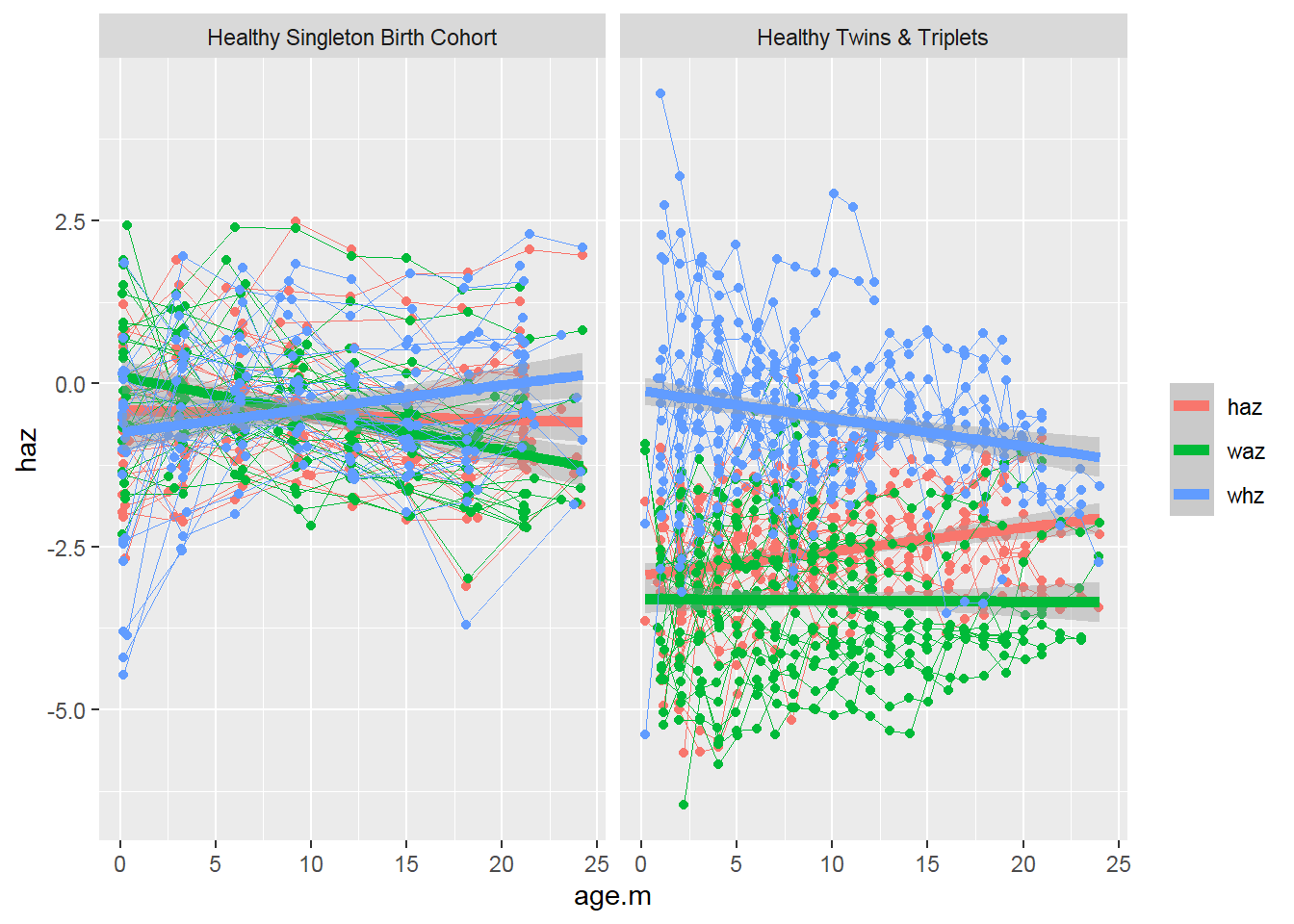
All-in-one spaghetti plot by cohort for each index and each plot for each index:
p1<-sh2 %>%
filter(!is.na(haz) & !is.na(waz) & !is.na(whz)) %>%
ggplot()+
geom_point(aes(x = age.m, y = haz, group = child.id, colour=cohort))+
geom_line(aes(x = age.m, y = haz, group = child.id, colour=cohort),size=0.3)+
stat_smooth(aes(x = age.m, y = haz, colour=cohort), method = 'glm',size=2)+
theme(legend.position = "bottom")
p2<-sh2 %>%
filter(!is.na(haz) & !is.na(waz) & !is.na(whz)) %>%
ggplot()+
geom_point(aes(x = age.m, y = waz, group = child.id, colour=cohort))+
geom_line(aes(x = age.m, y = waz, group = child.id, colour=cohort),size=0.3)+
stat_smooth(aes(x = age.m, y = waz, colour=cohort), method = 'glm',size=2)+
theme(legend.position = "bottom")
p3<-sh2 %>%
filter(!is.na(haz) & !is.na(waz) & !is.na(whz)) %>%
ggplot()+
geom_point(aes(x = age.m, y = whz, group = child.id, colour=cohort))+
geom_line(aes(x = age.m, y = whz, group = child.id, colour=cohort),size=0.3)+
stat_smooth(aes(x = age.m, y = whz, colour=cohort), method = 'glm',size=2)+
theme(legend.position = "bottom")
# Combine 3 plots into 1 plot
#install.packages("ggpubr")
library(ggpubr)
ggarrange(p1, p2, p3,nrow=1, common.legend = TRUE, legend="bottom")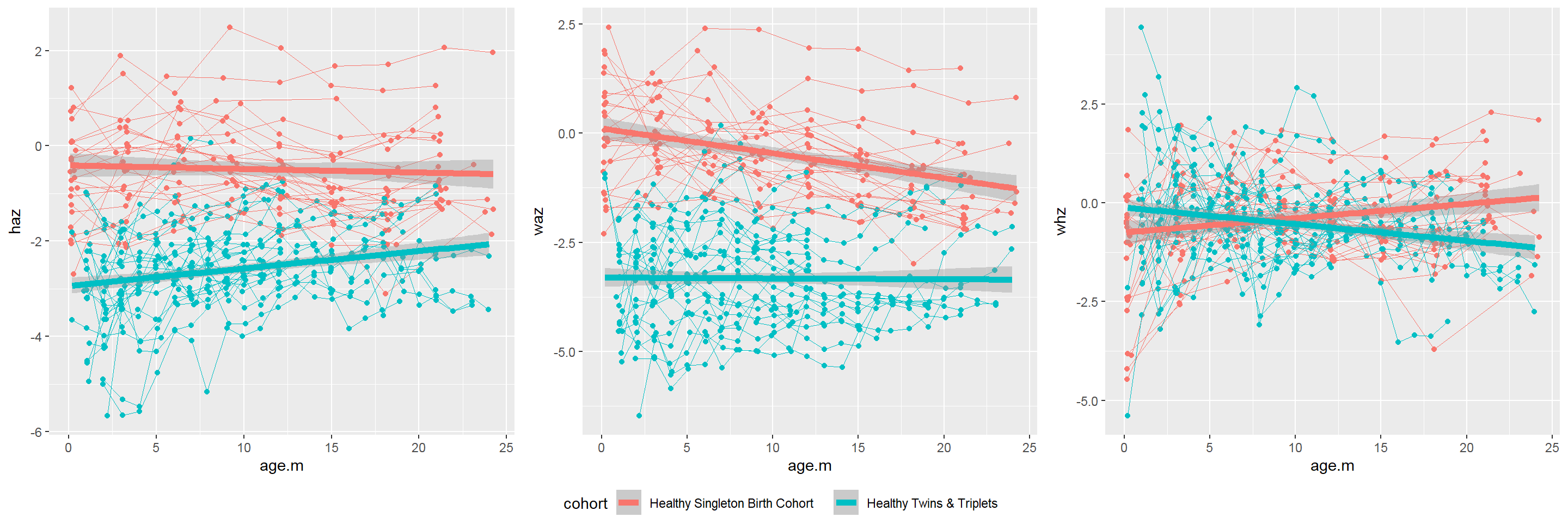
2.12 Reference materials
2.12.2 Datasets
- The datasets used in some examples of this book are of Bangladeshi infants published by Subramanian et al, Nature 2014. These datasets may be downloaded from Gordon’s lab website, or from my github repo.