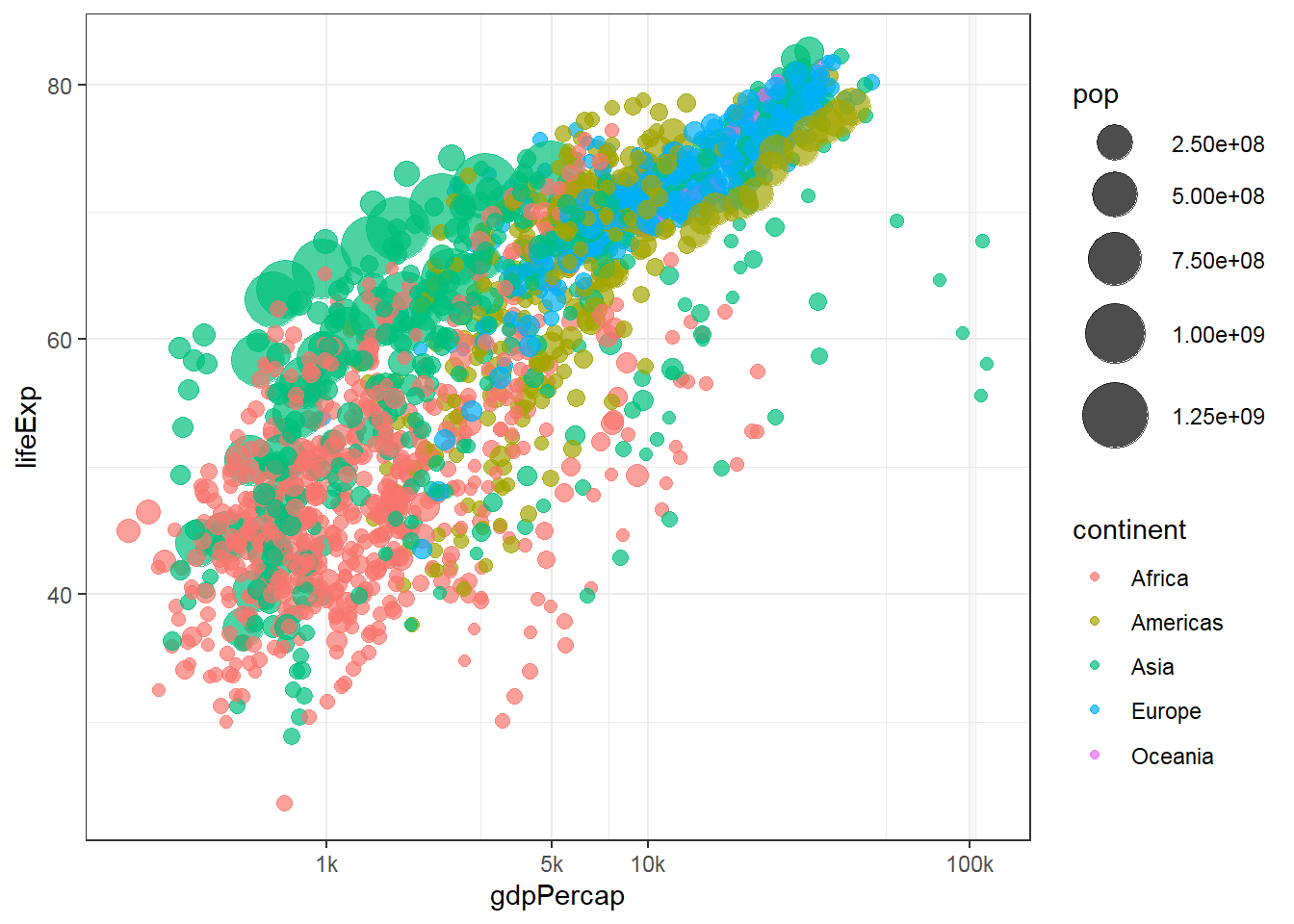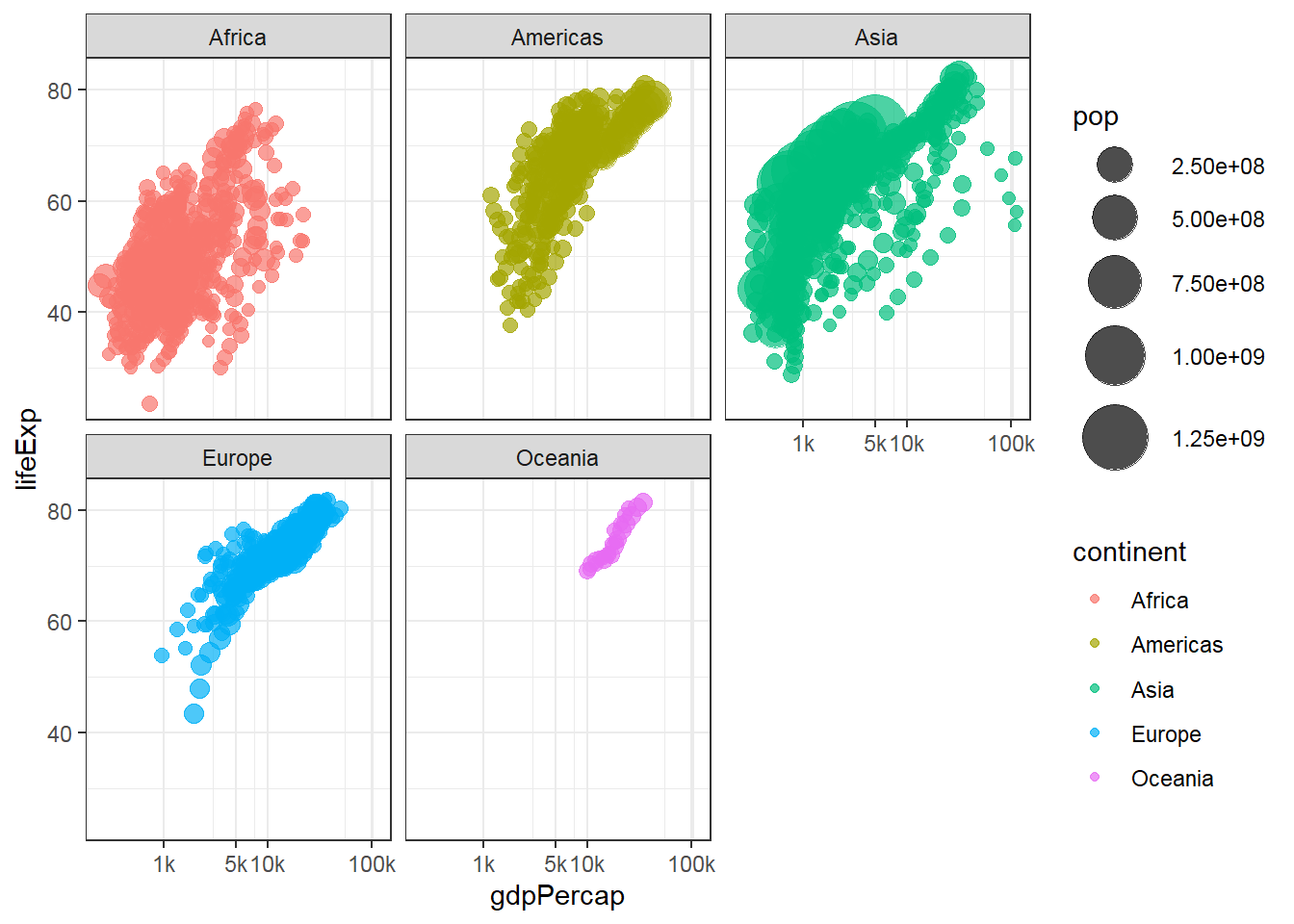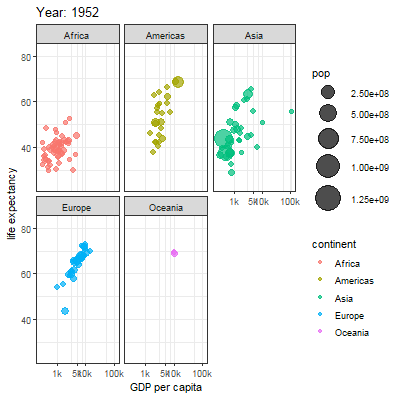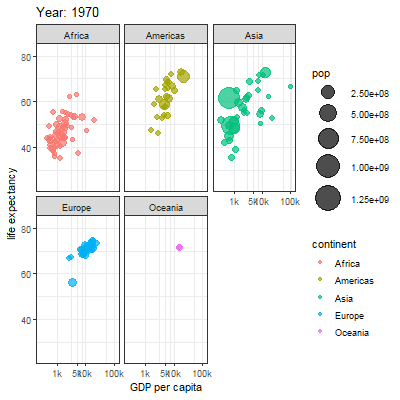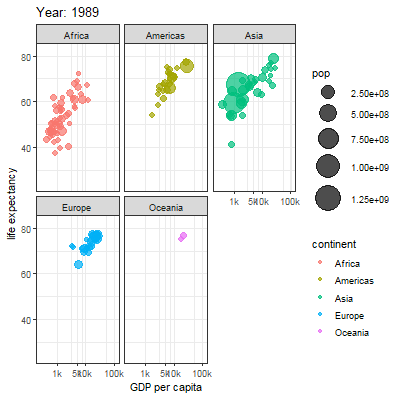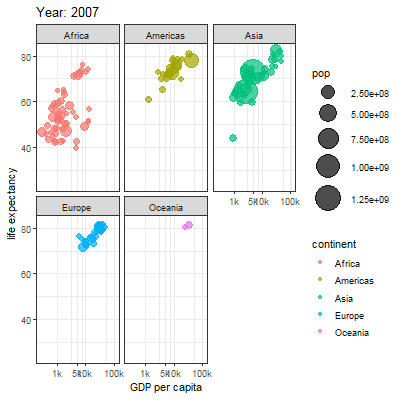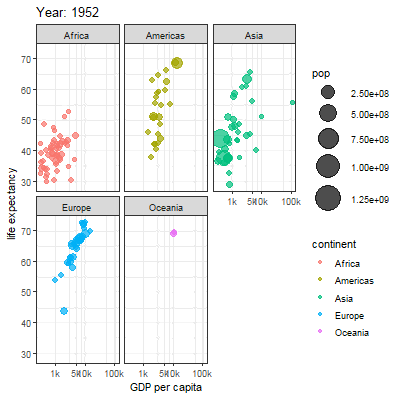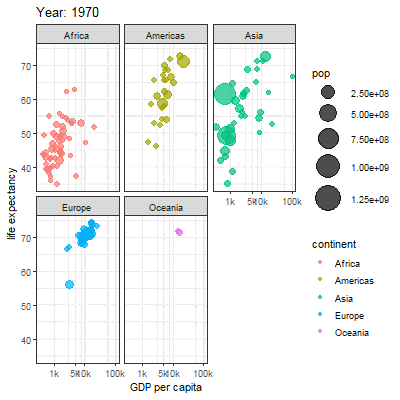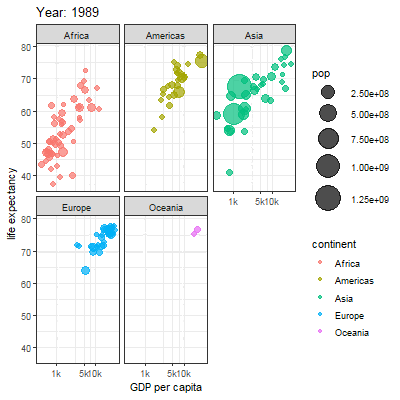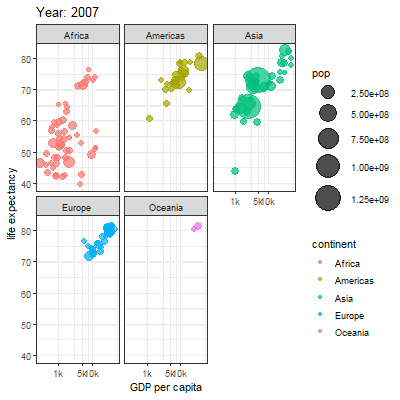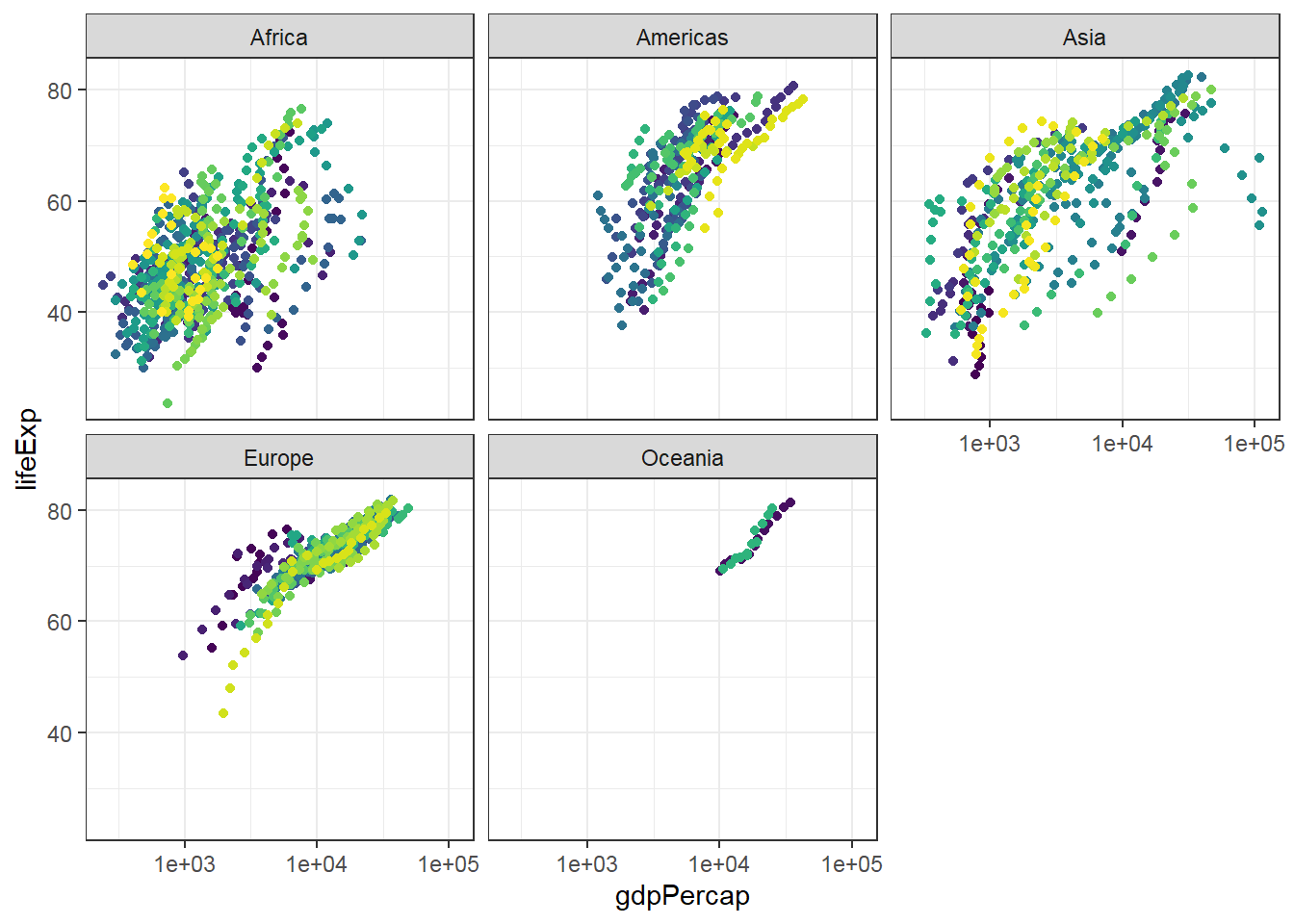
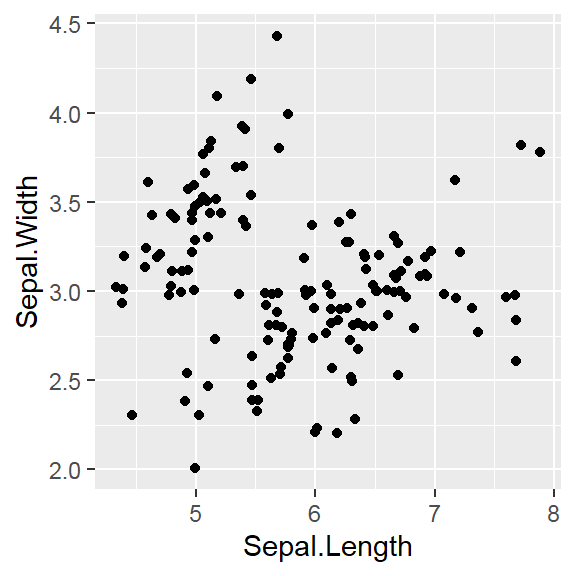
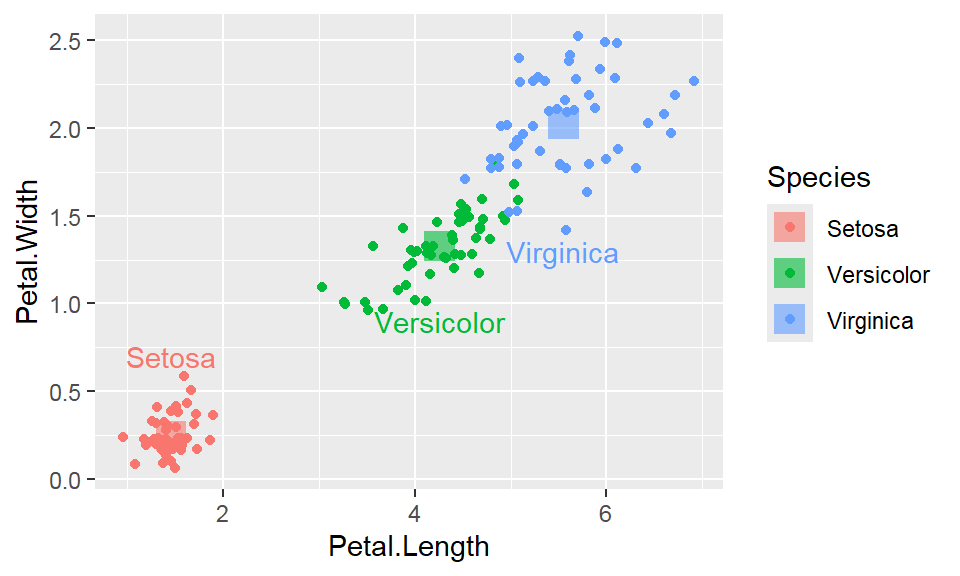
 They are discrete variables.
They are discrete variables.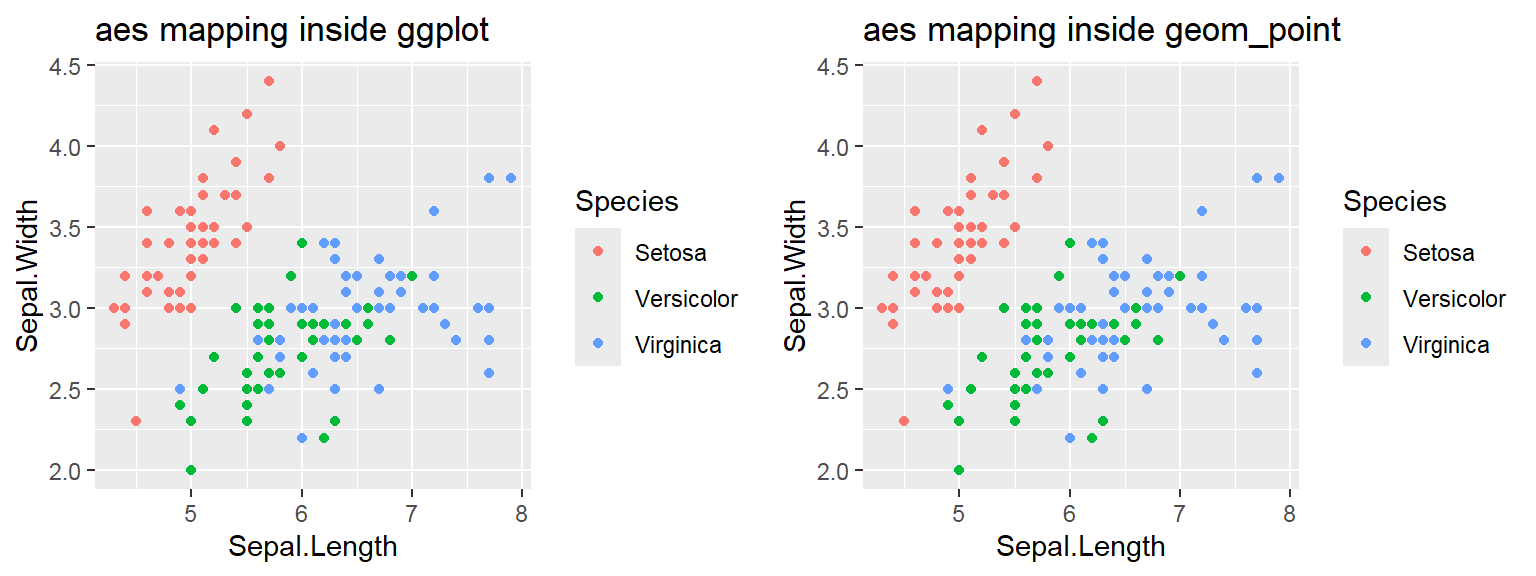
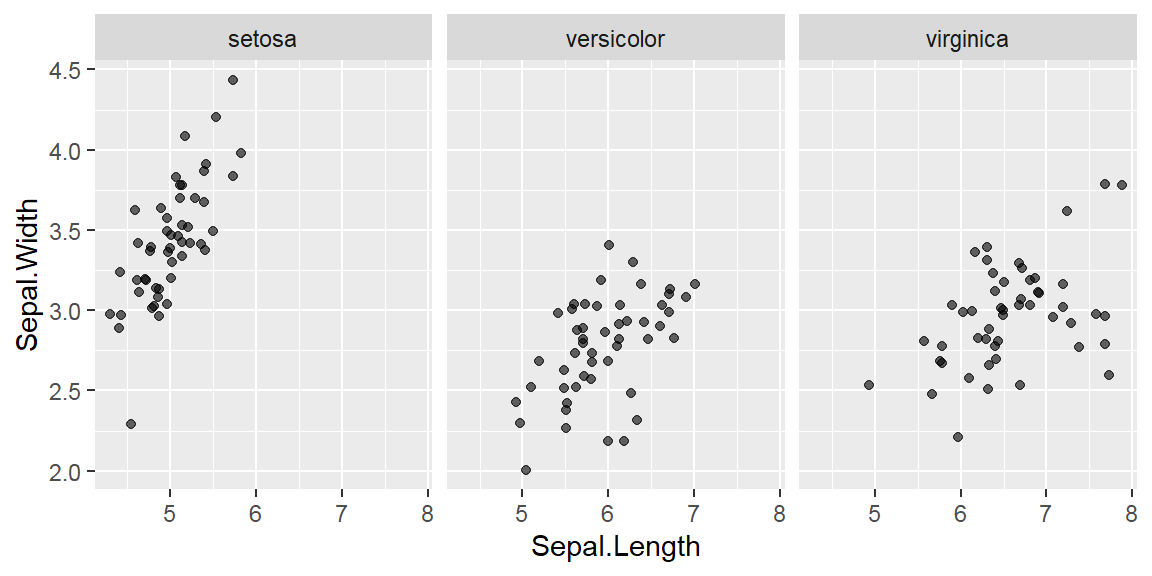
 The color attribute is set to “red”
Attribute is set inside geom_*() .
The color attribute is set to “red”
Attribute is set inside geom_*() .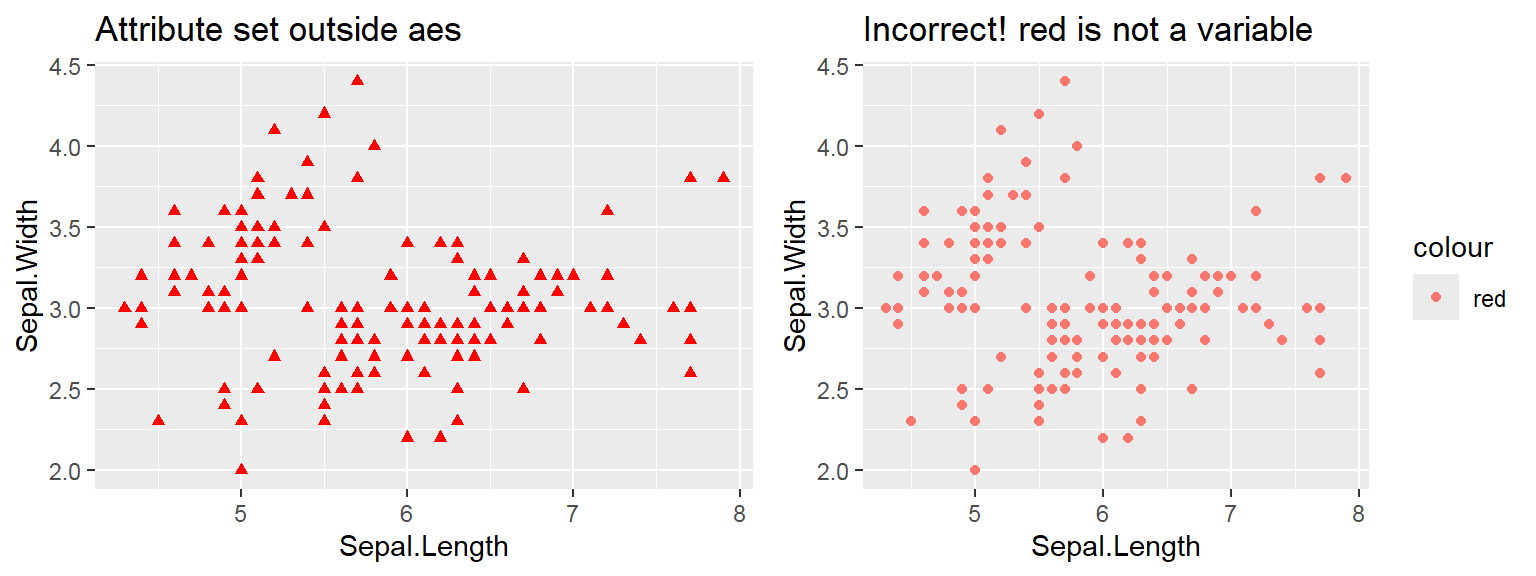
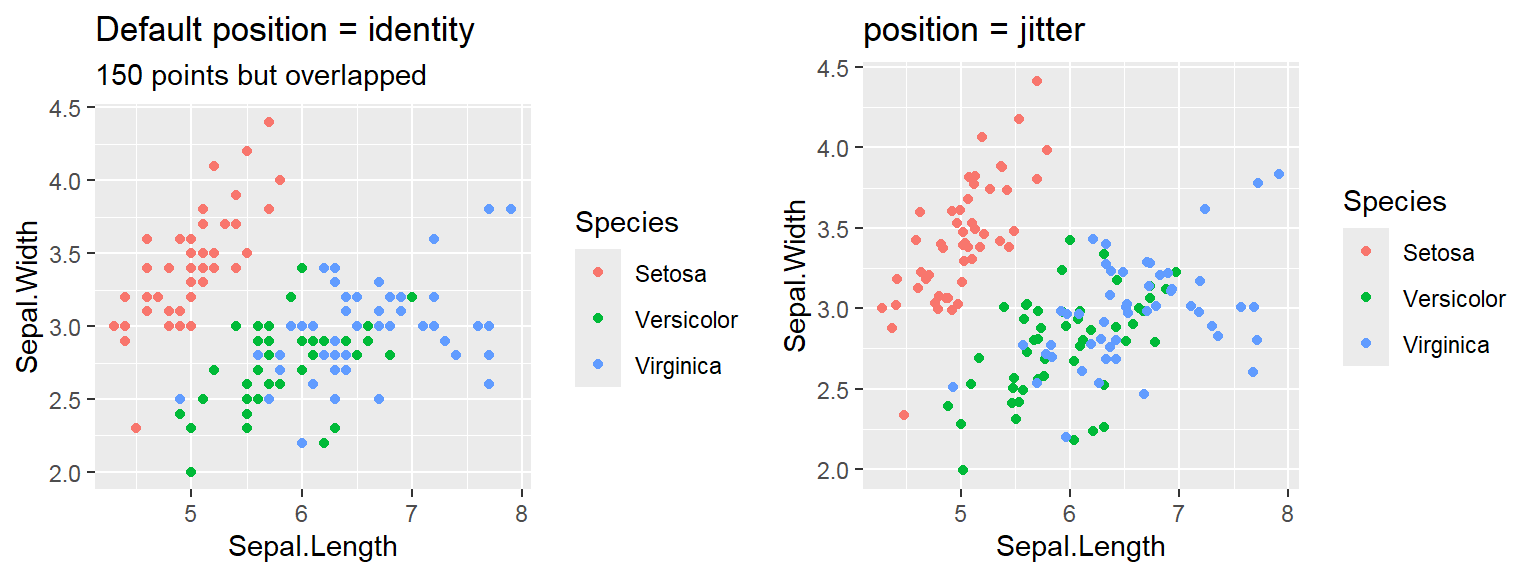
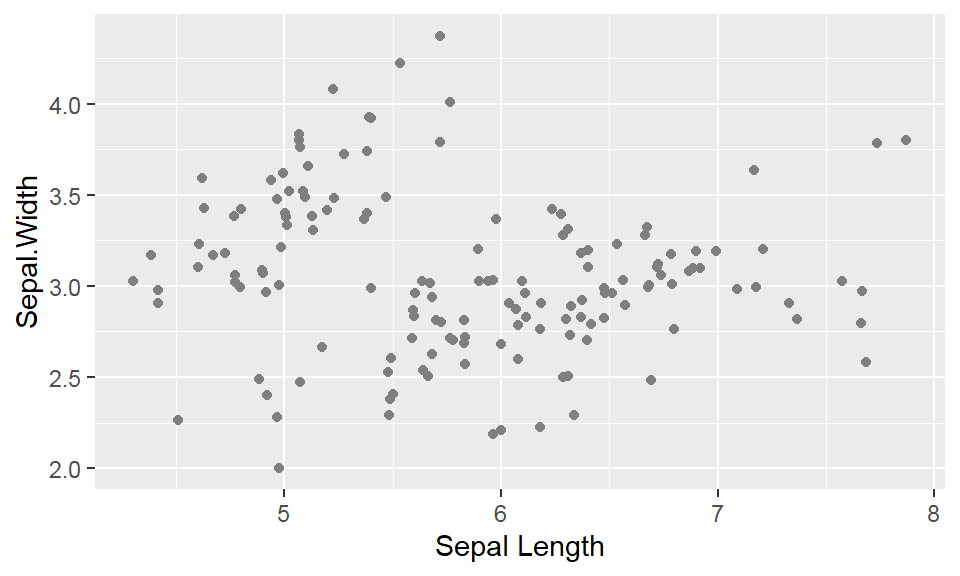
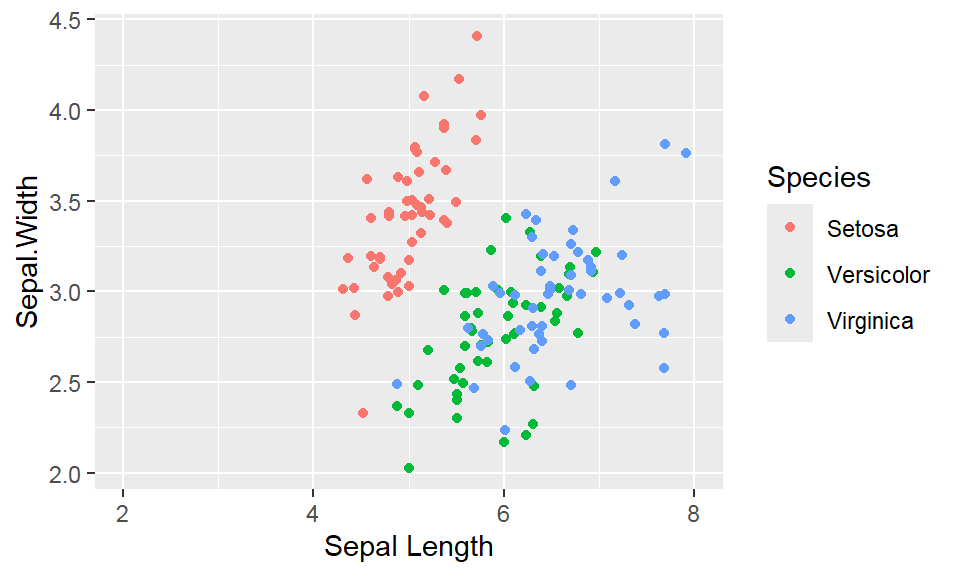
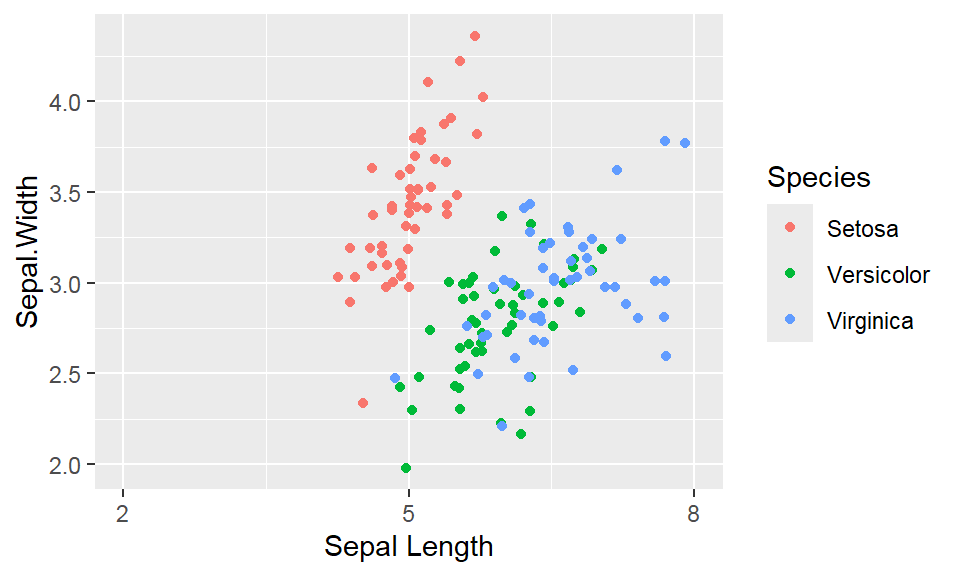
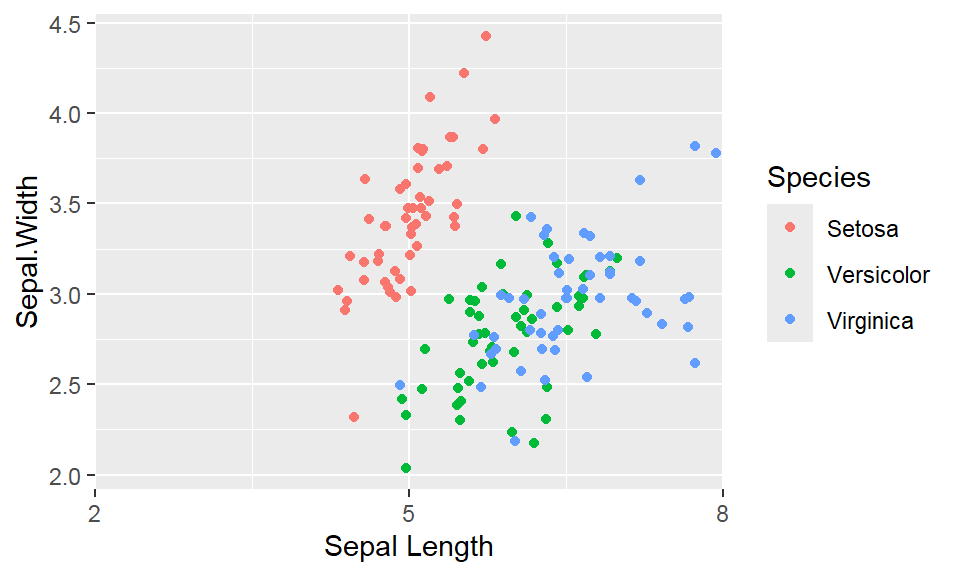
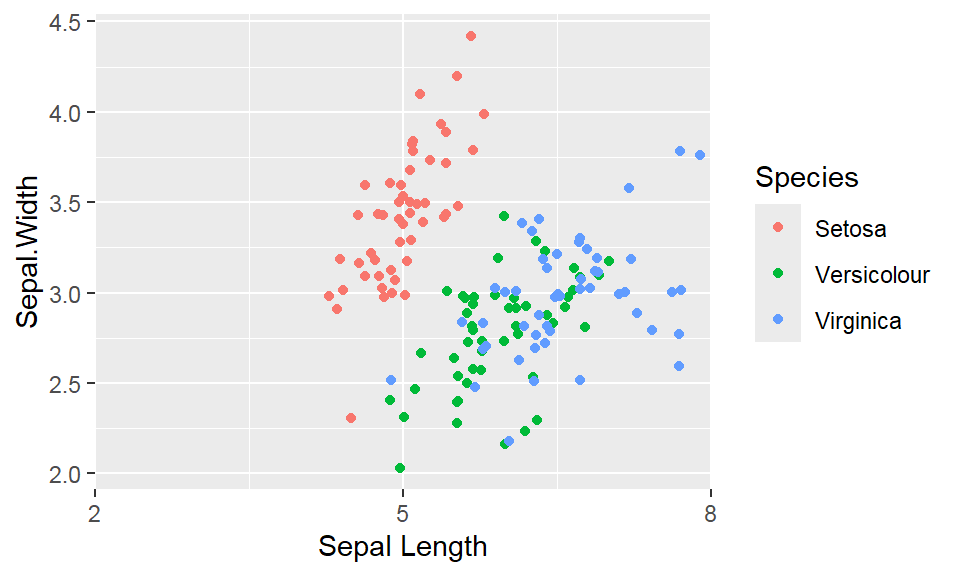
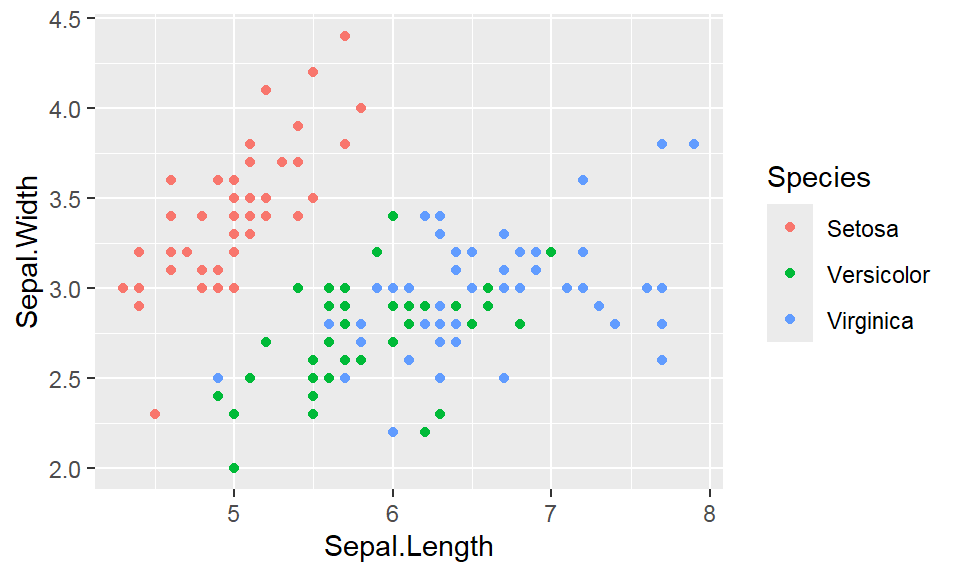
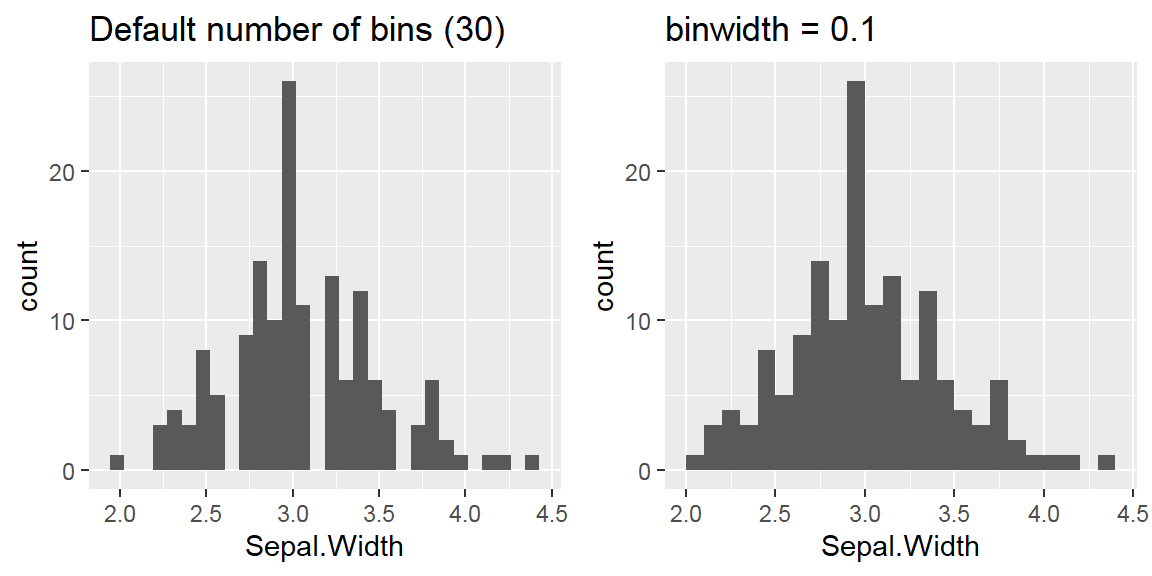
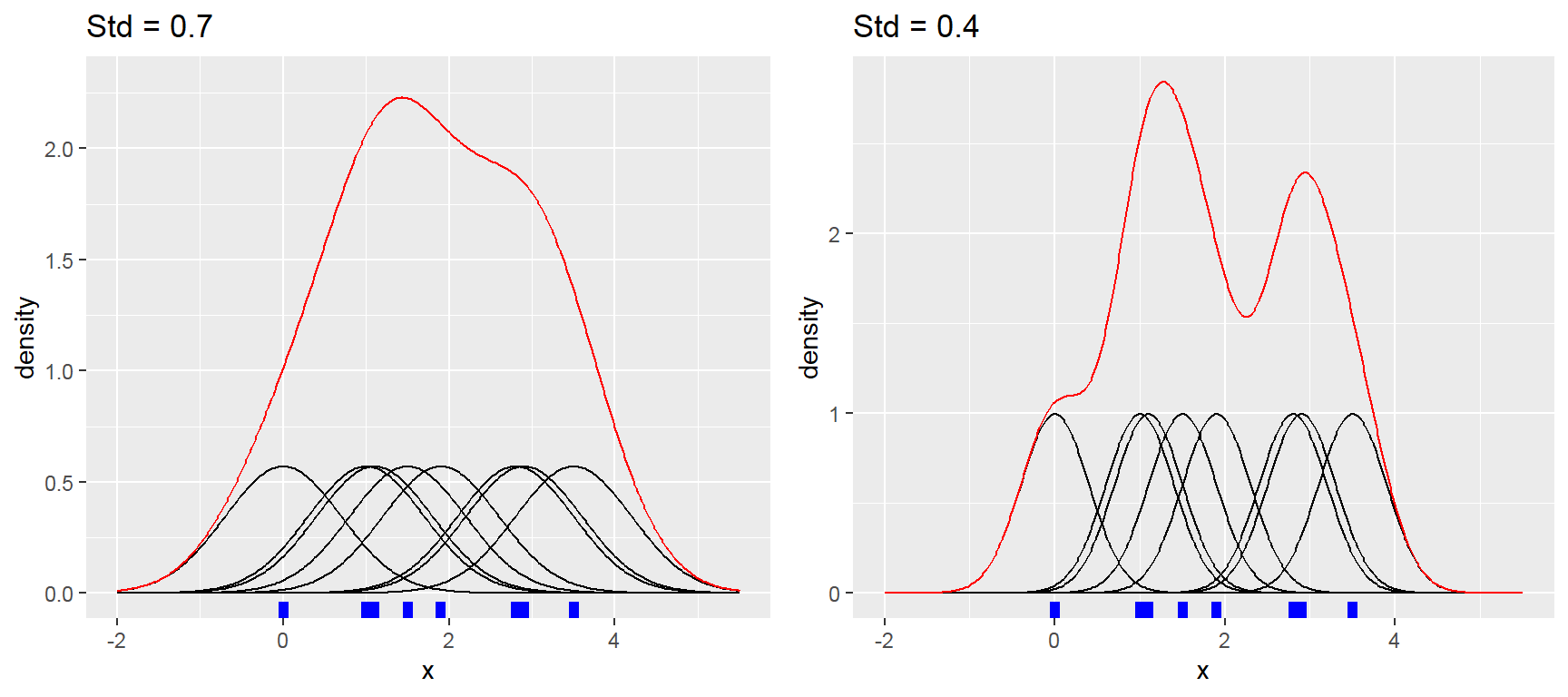
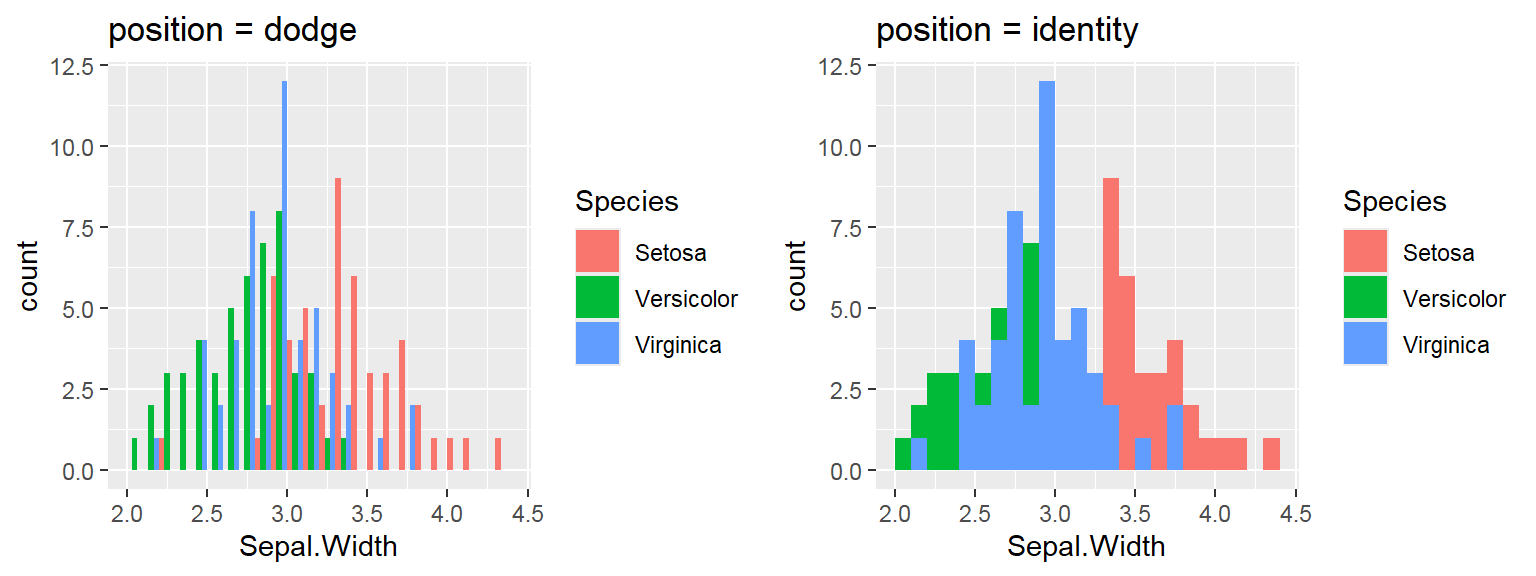
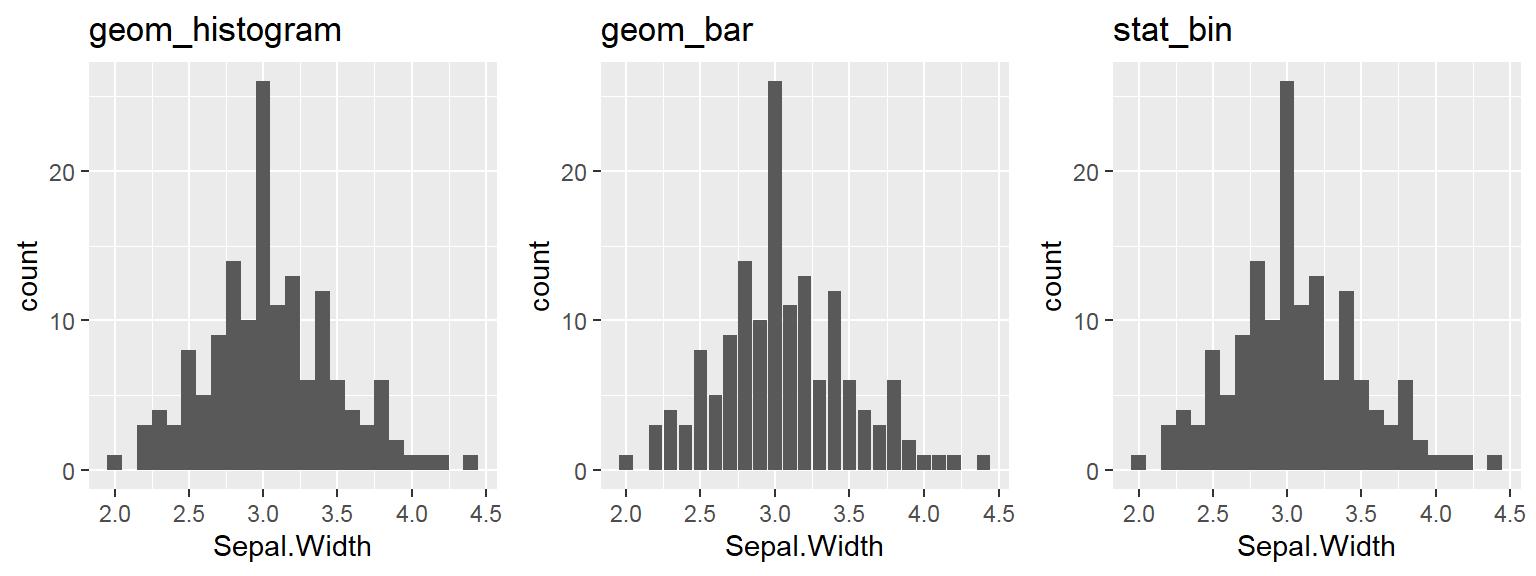
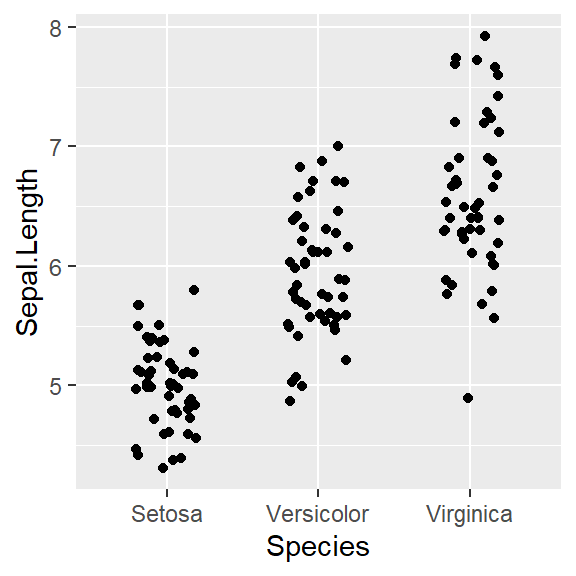
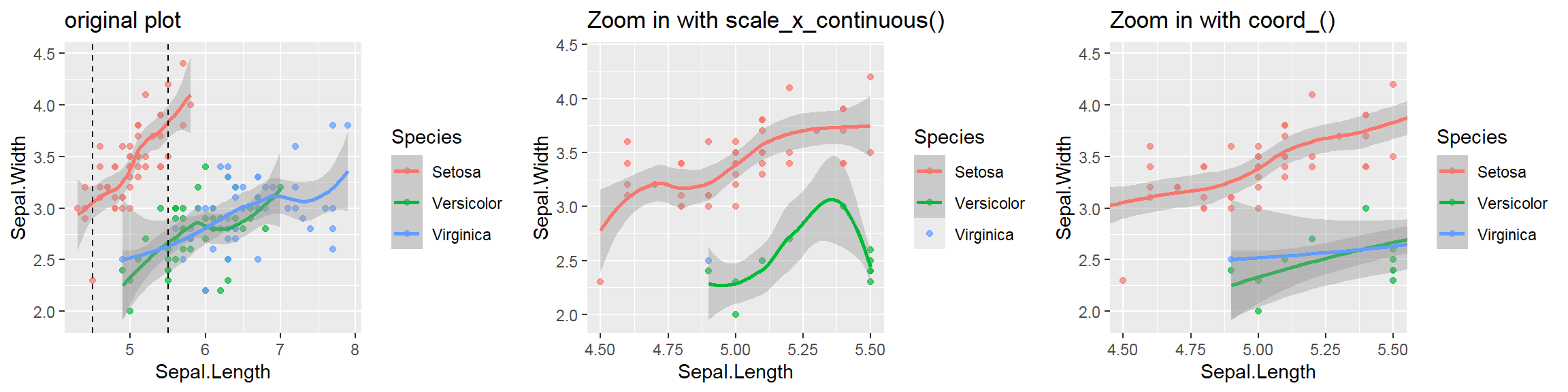
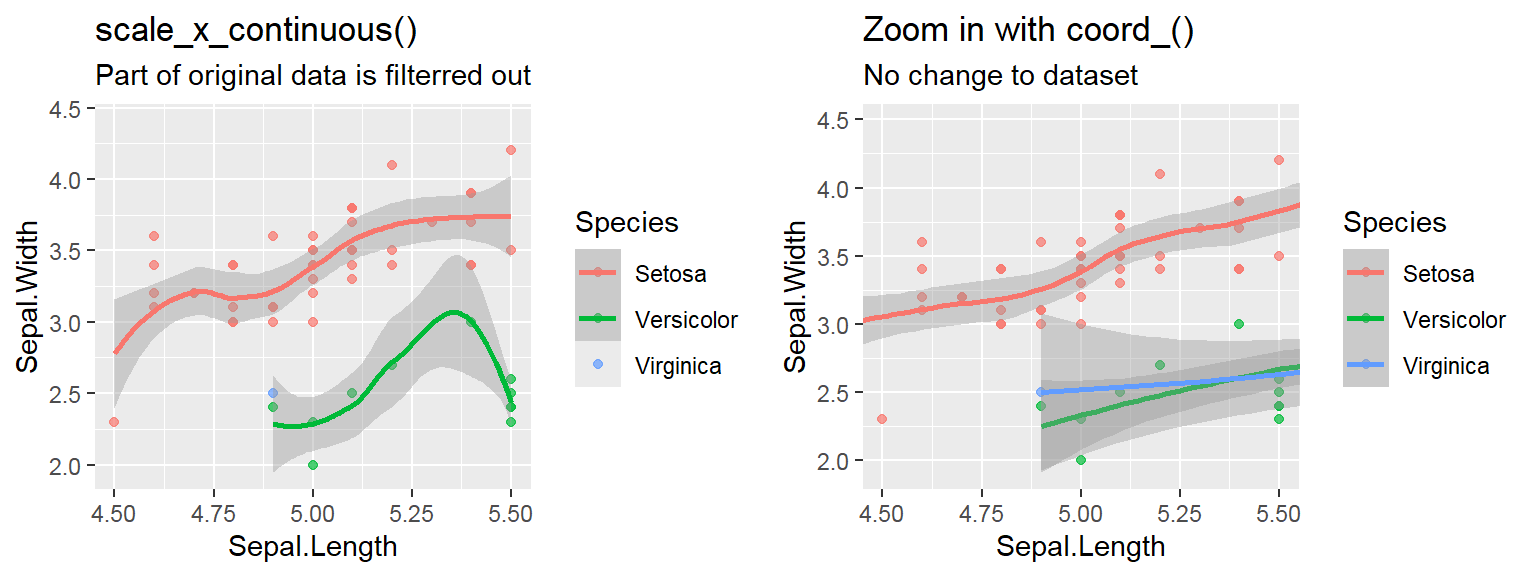
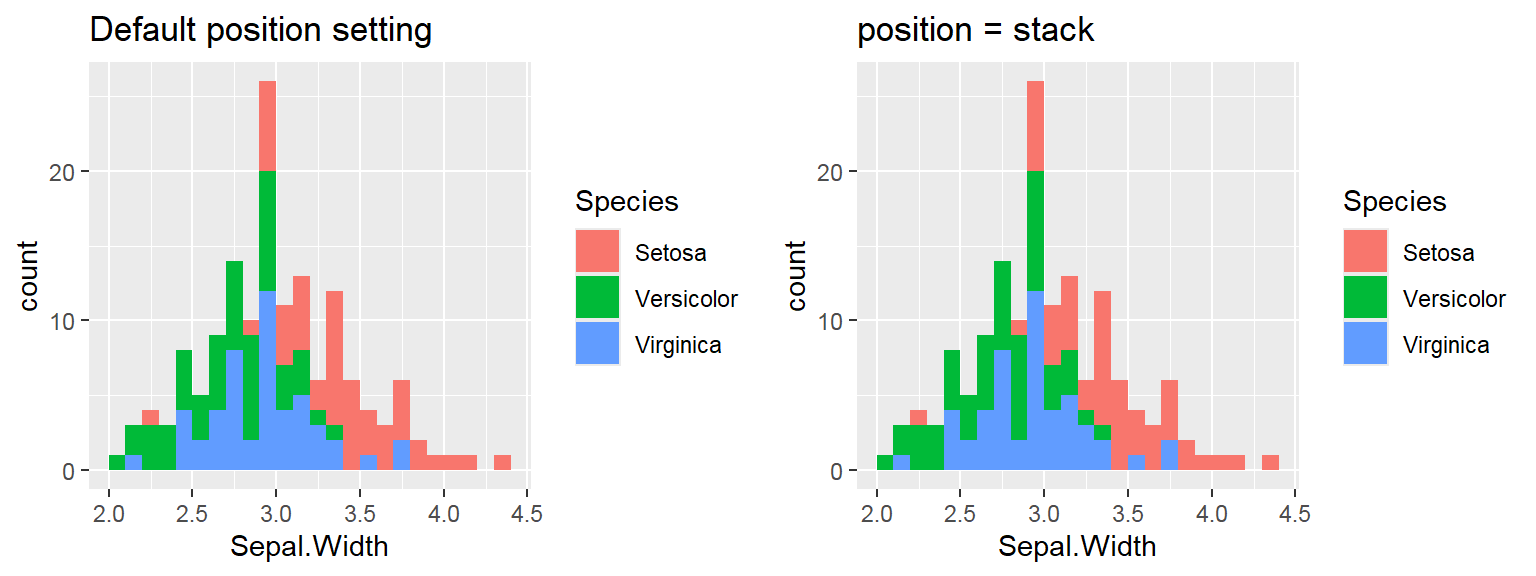 We cant say whether the histogram bars are stacked or overlapped onto each other
We cant say whether the histogram bars are stacked or overlapped onto each other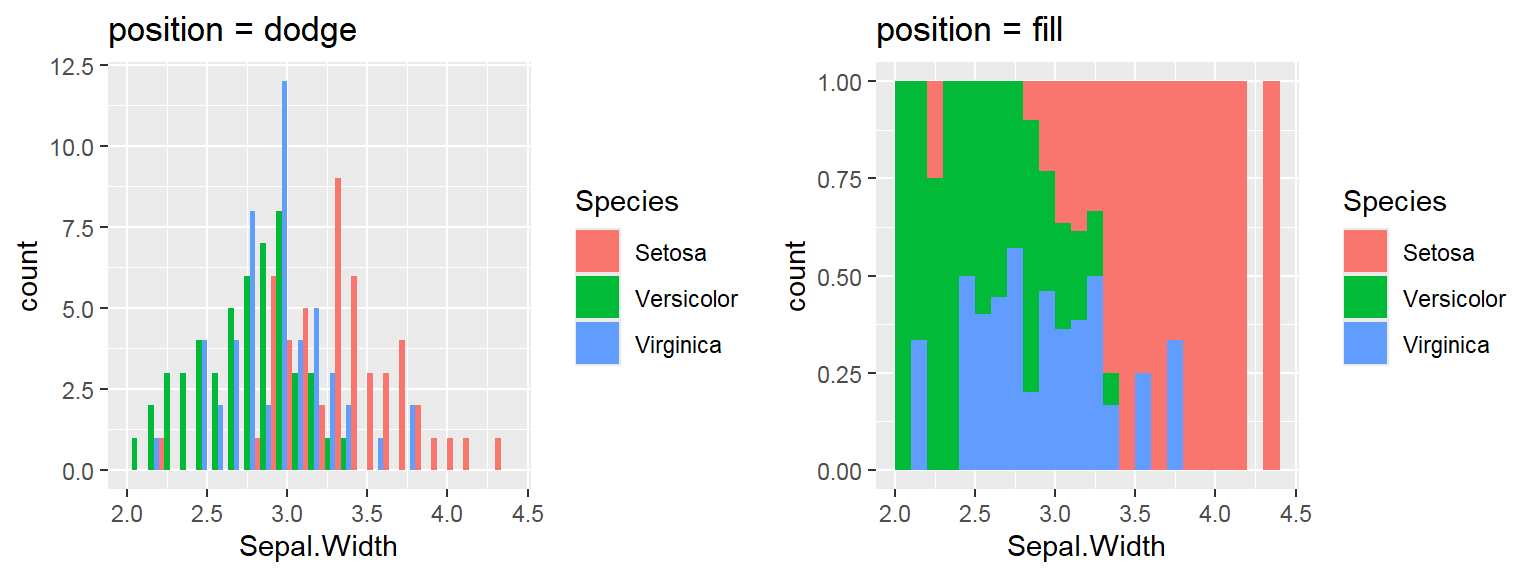
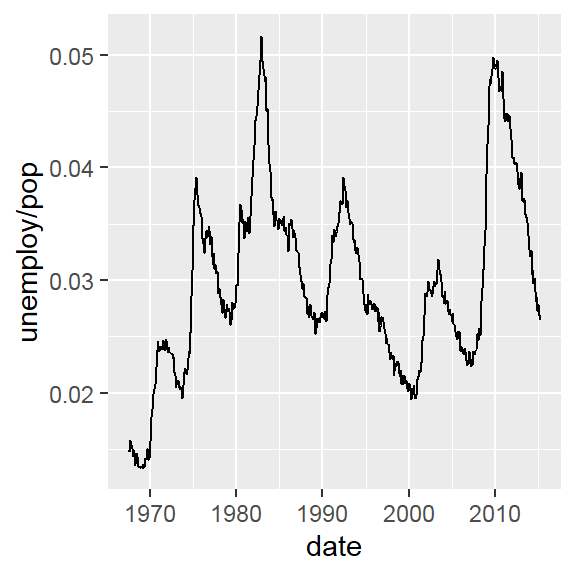
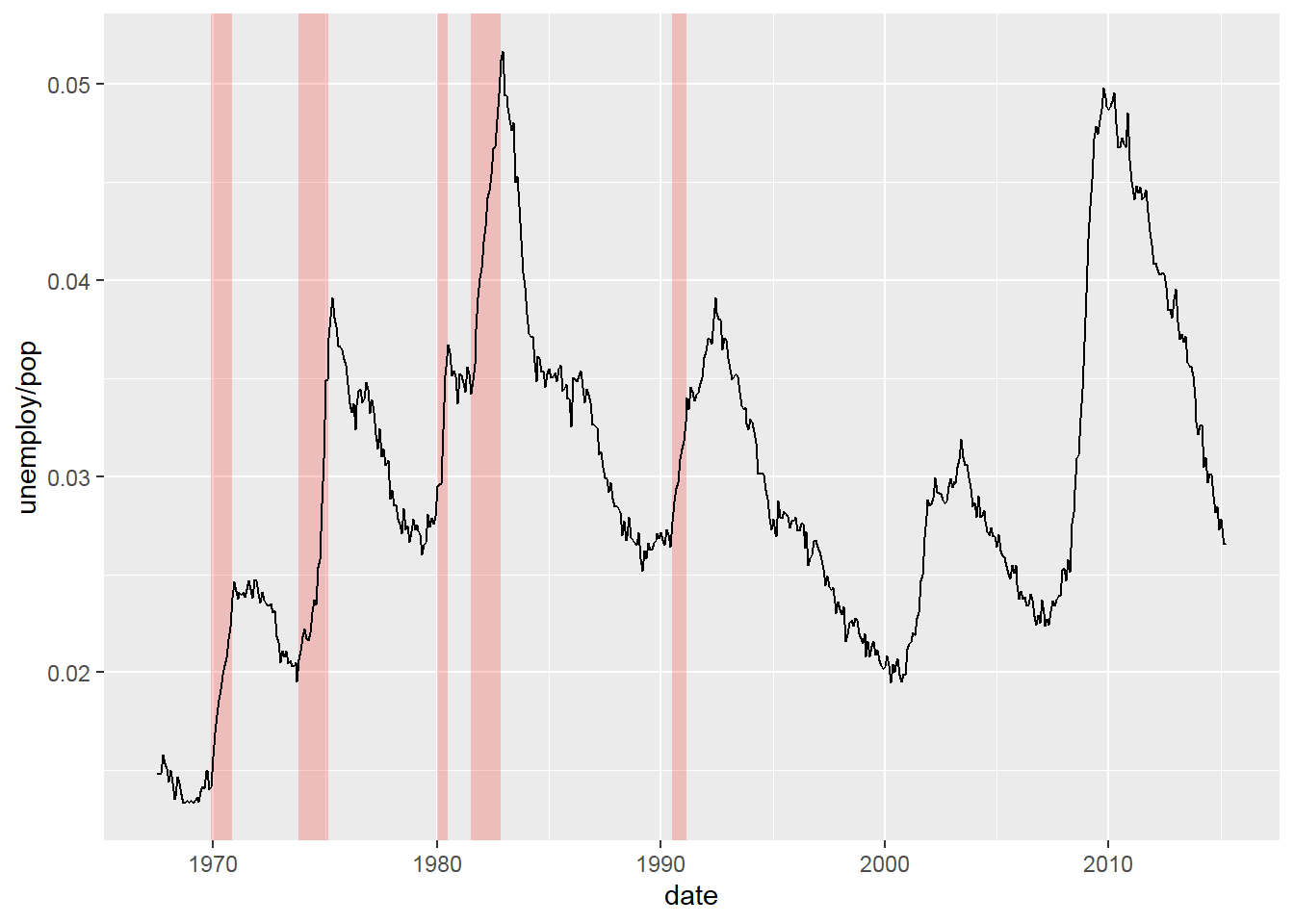
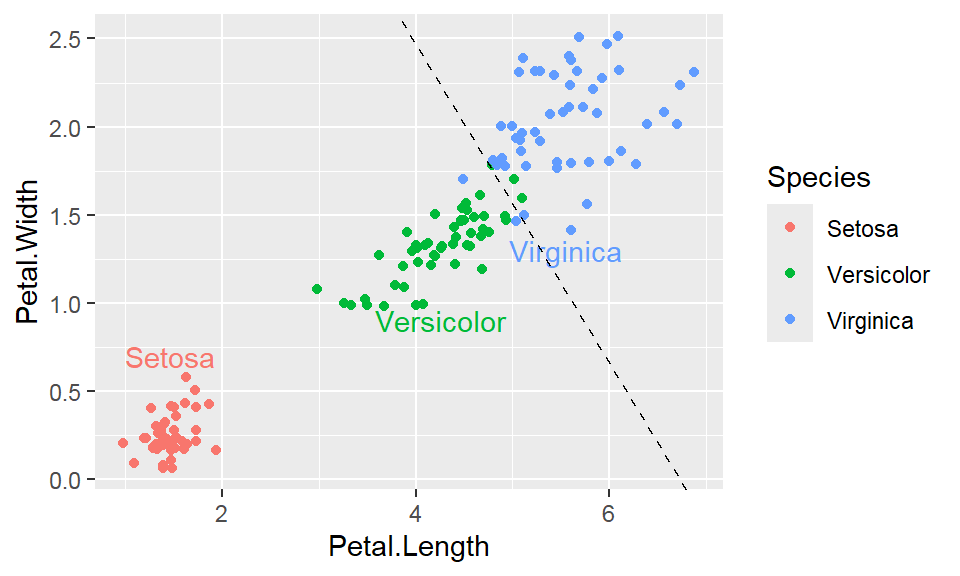
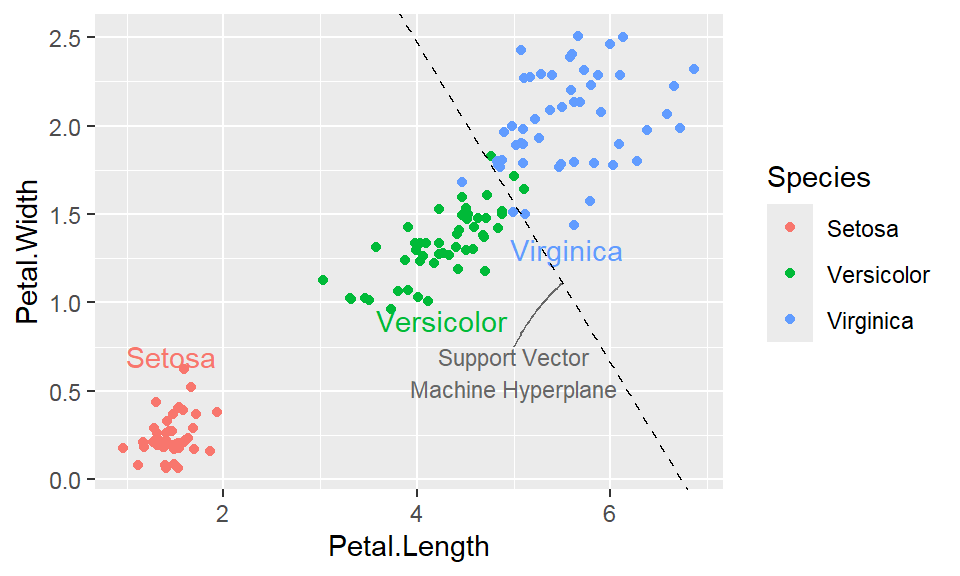
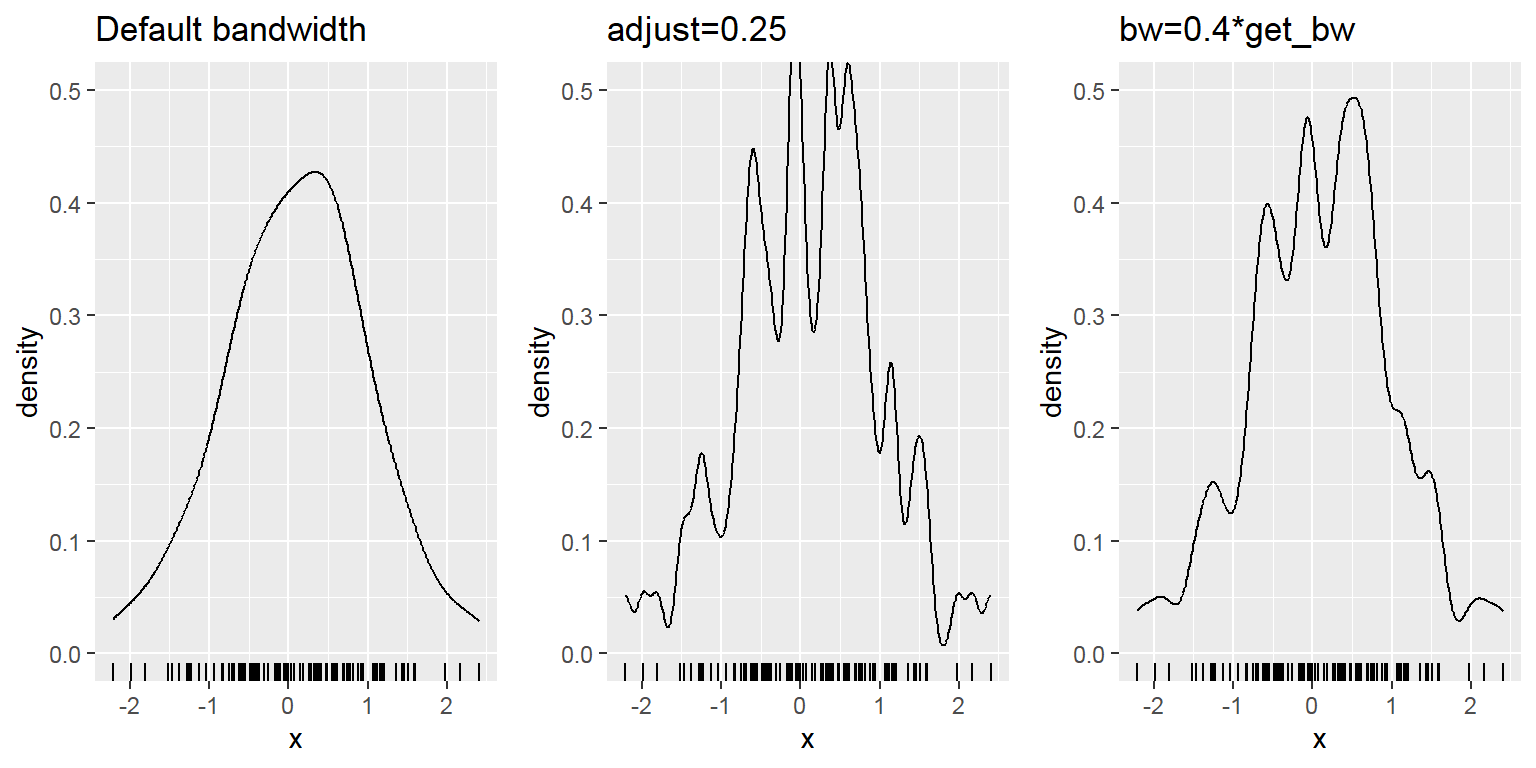
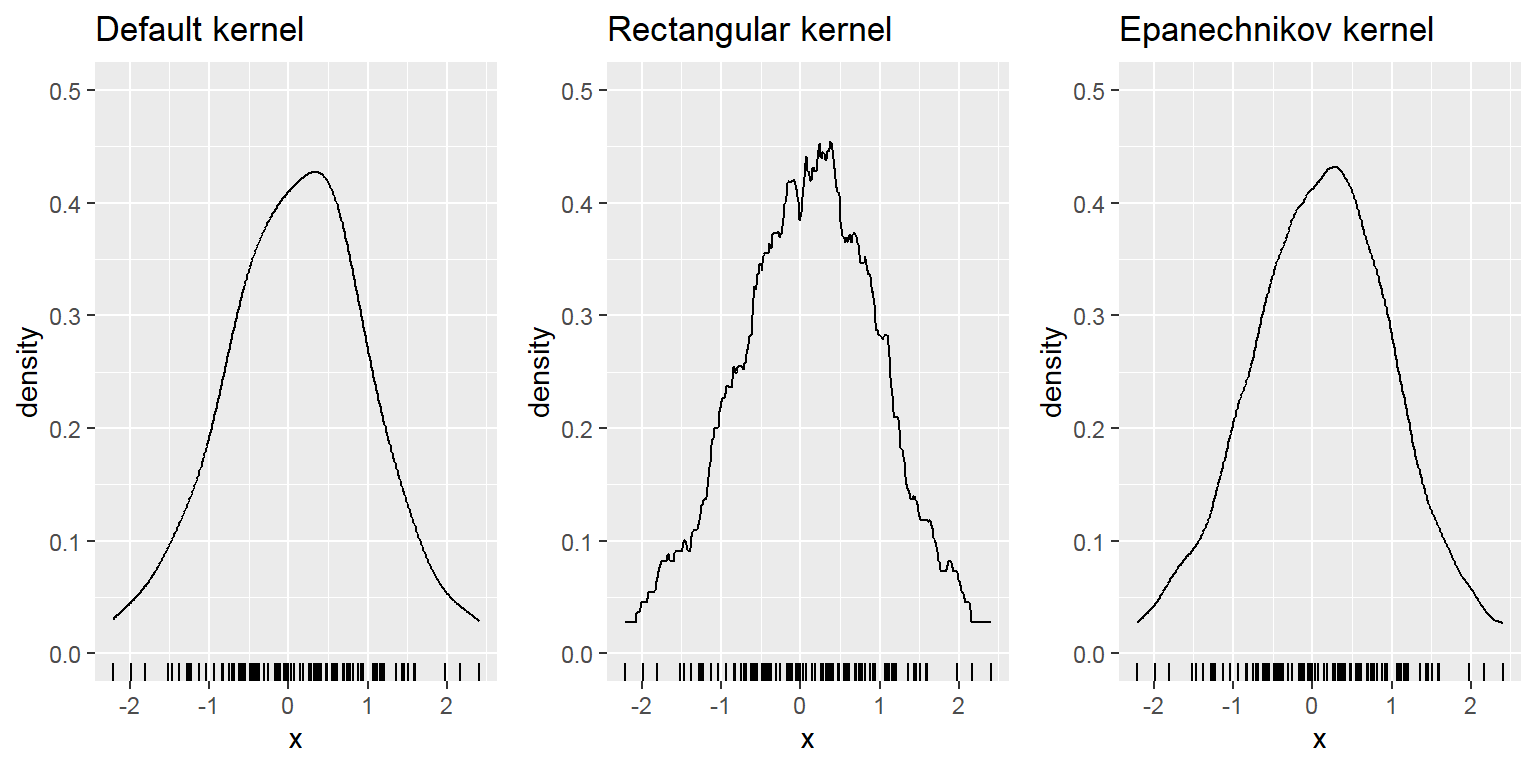
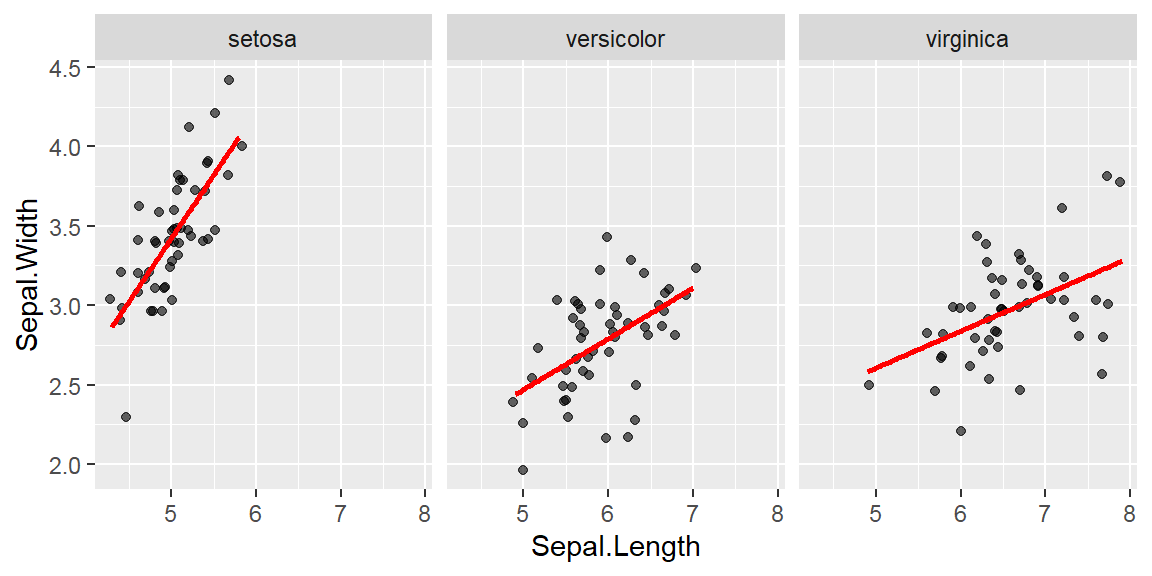
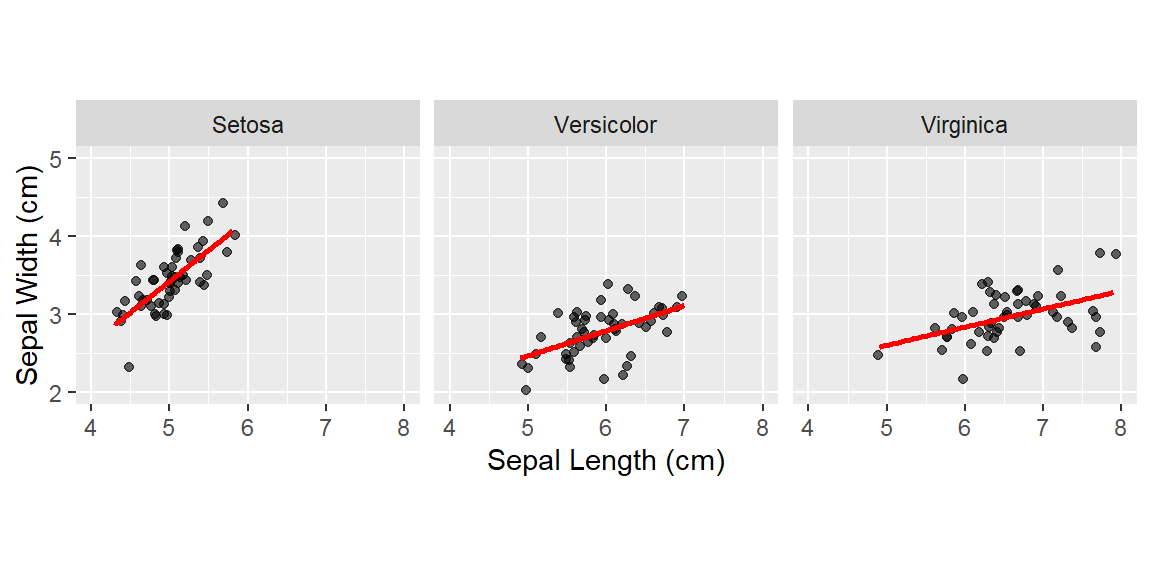
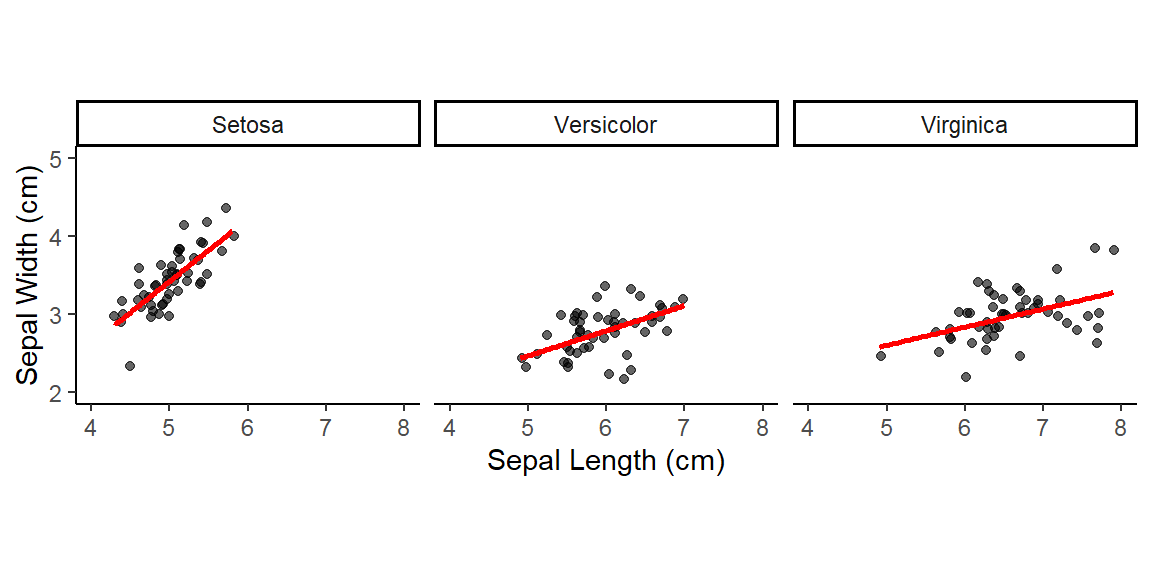
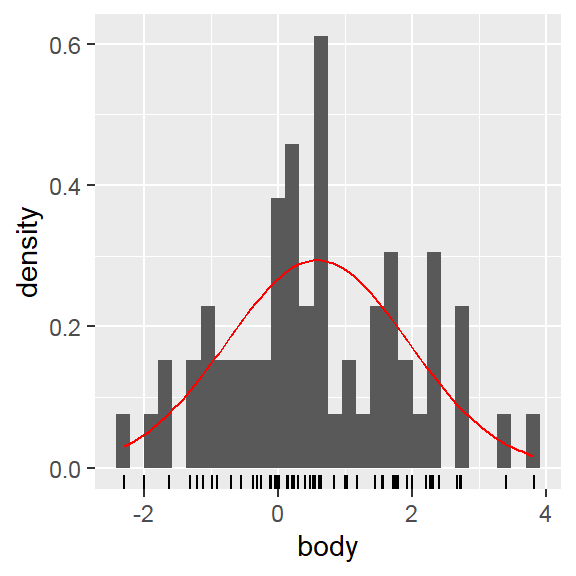
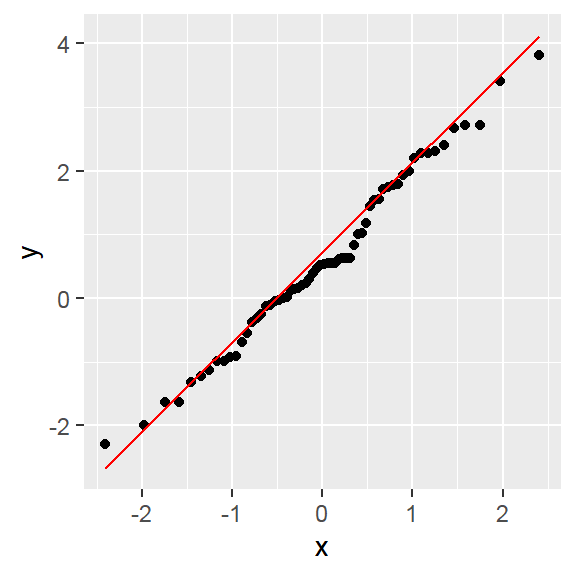
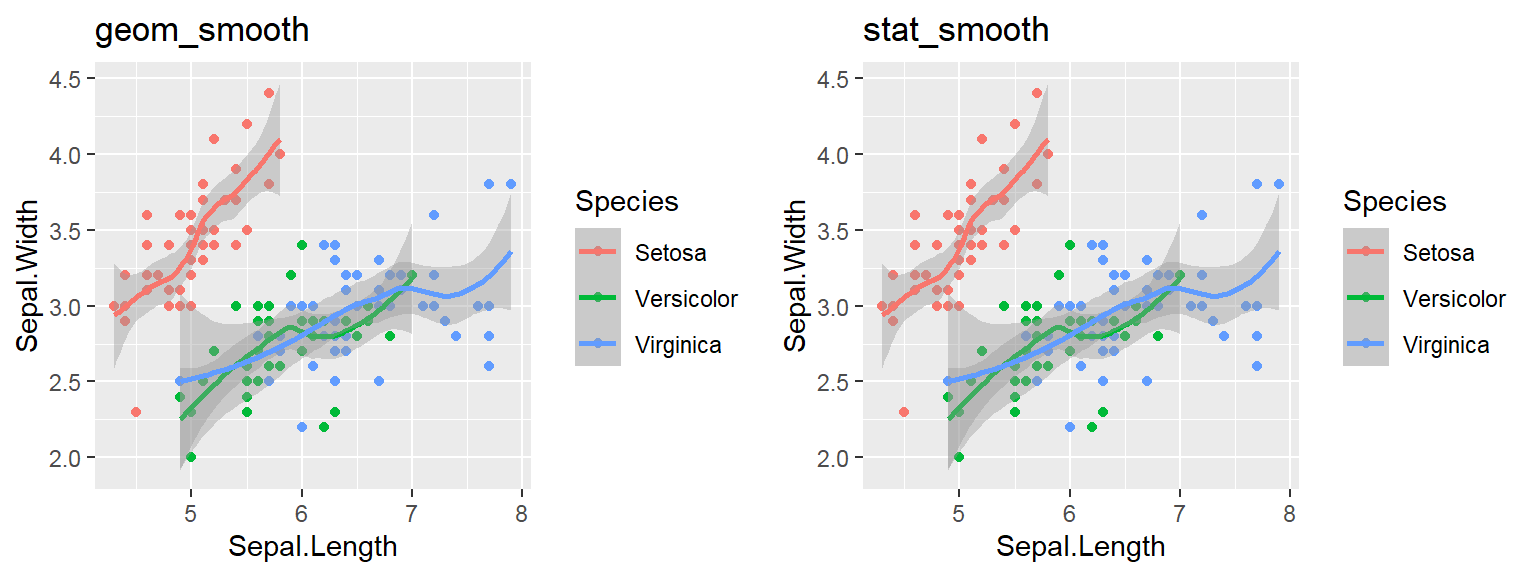 Note: By default, loess regression is used. It is a non-parametric methods where least squares regression is performed in localized subsets, and used when n < 1000. We can change smoothing method with the
Note: By default, loess regression is used. It is a non-parametric methods where least squares regression is performed in localized subsets, and used when n < 1000. We can change smoothing method with the 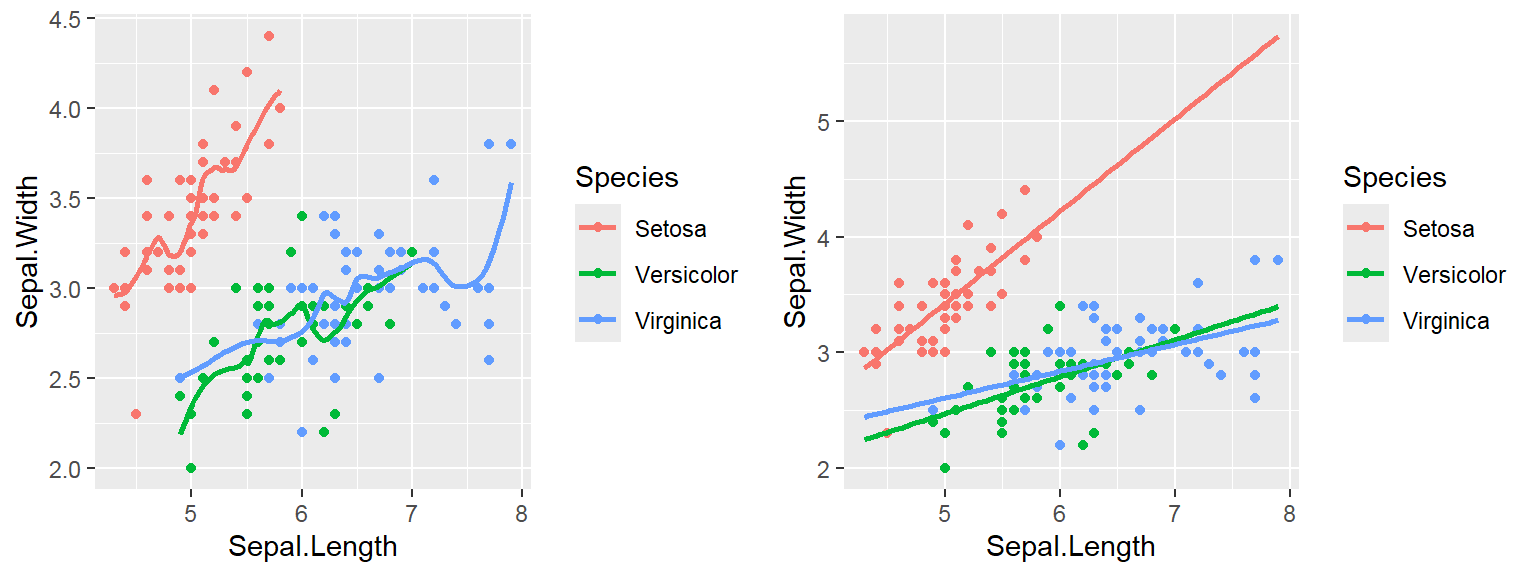
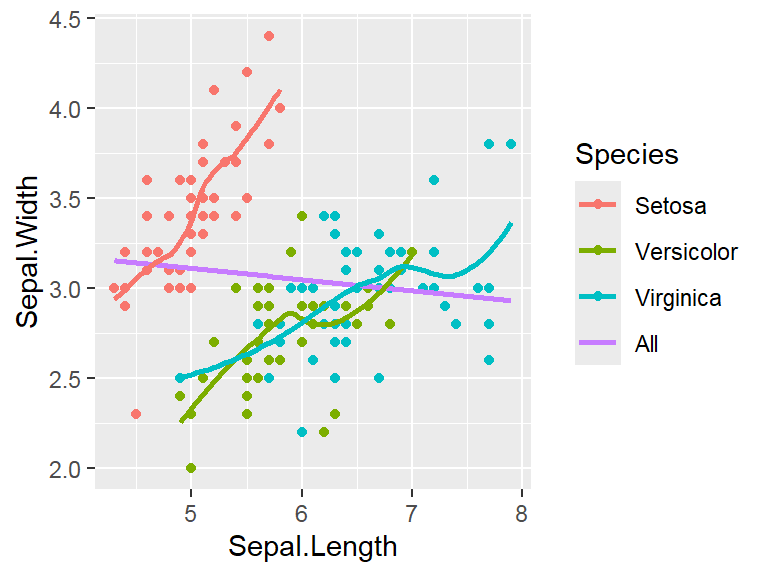
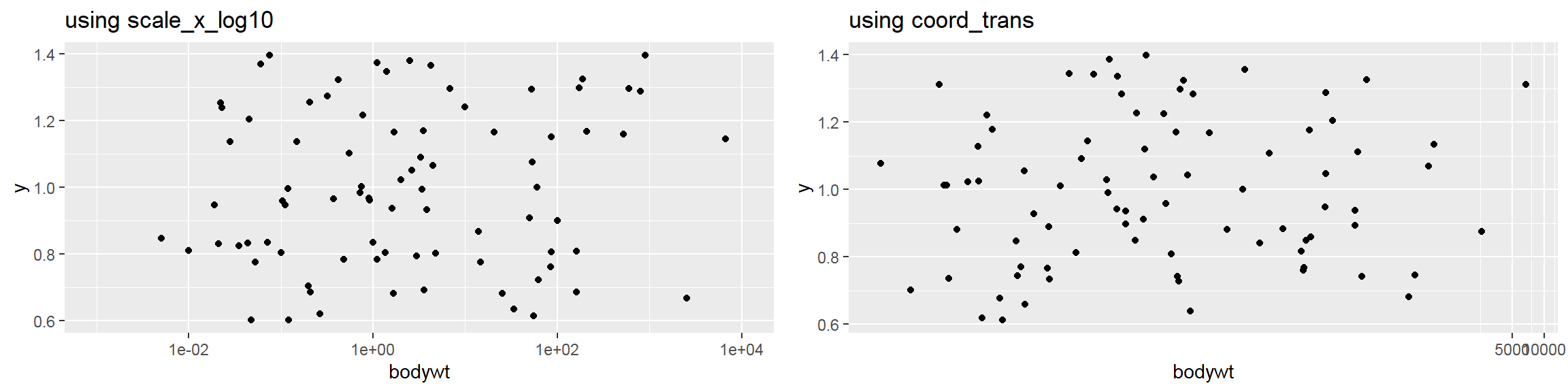
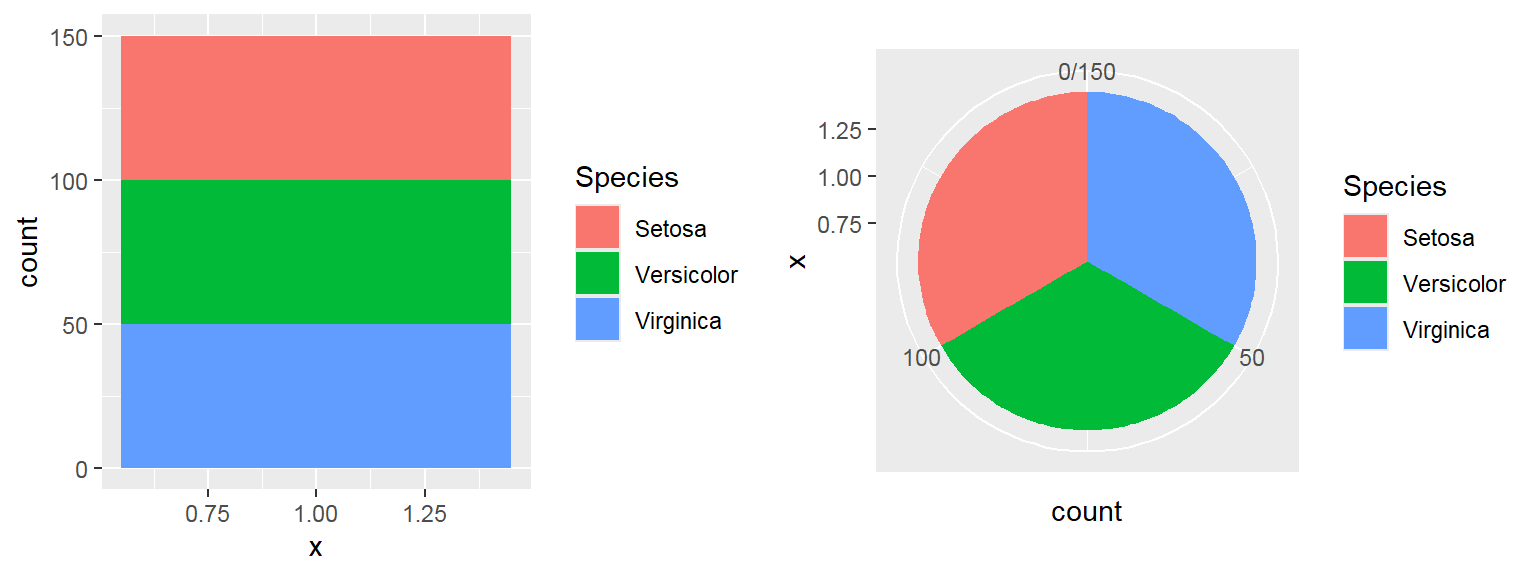

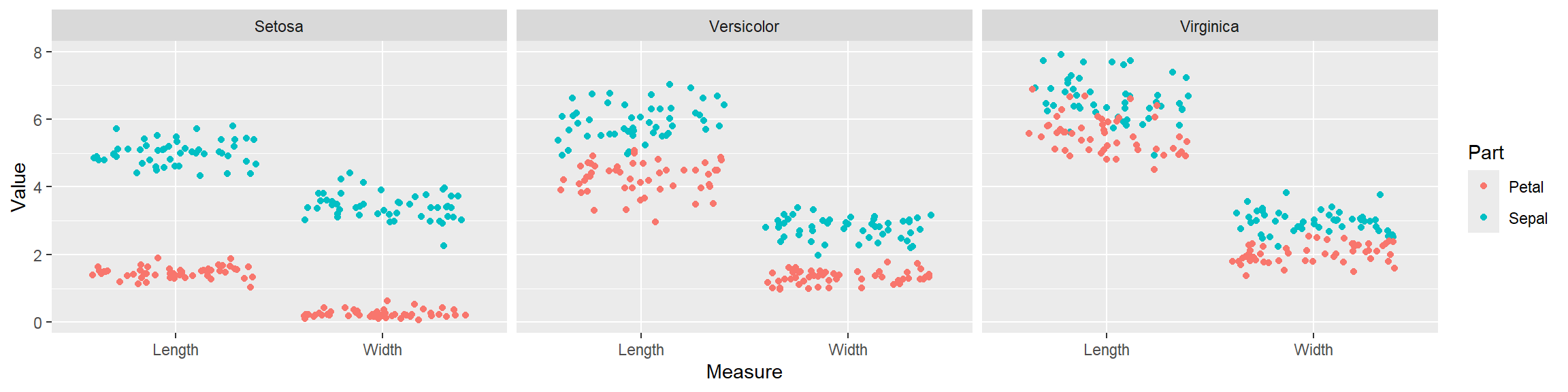
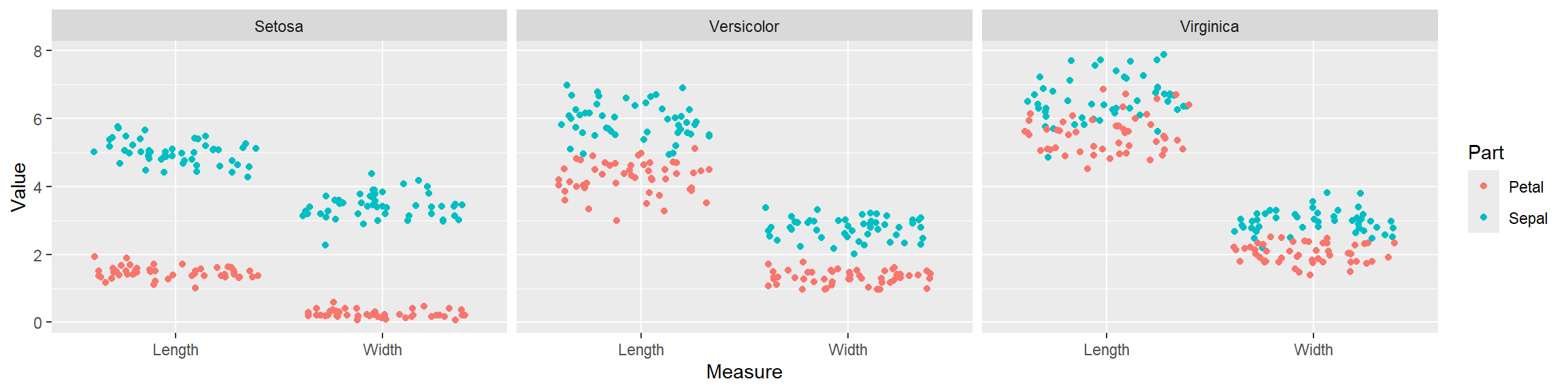
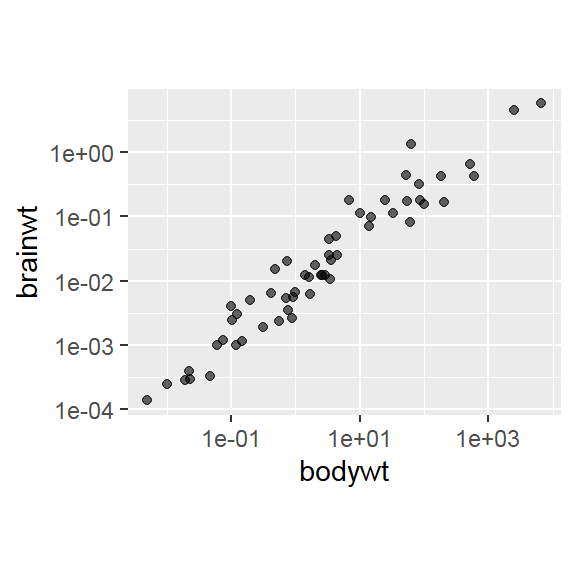
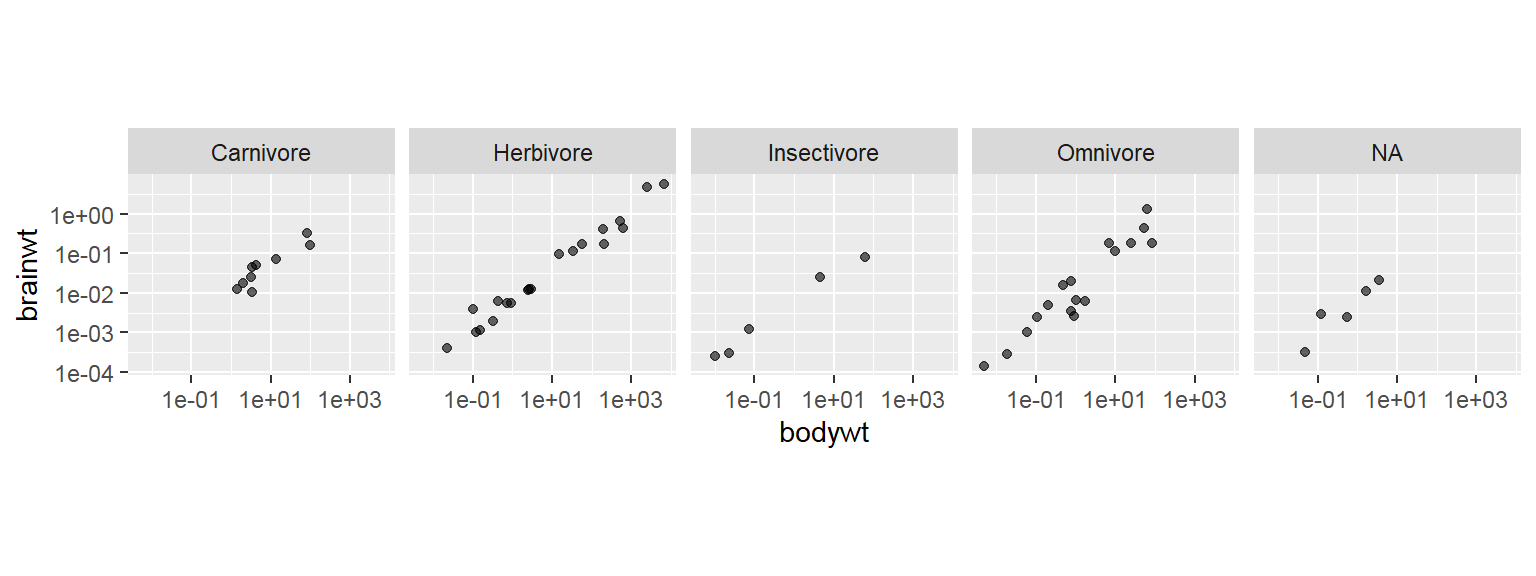
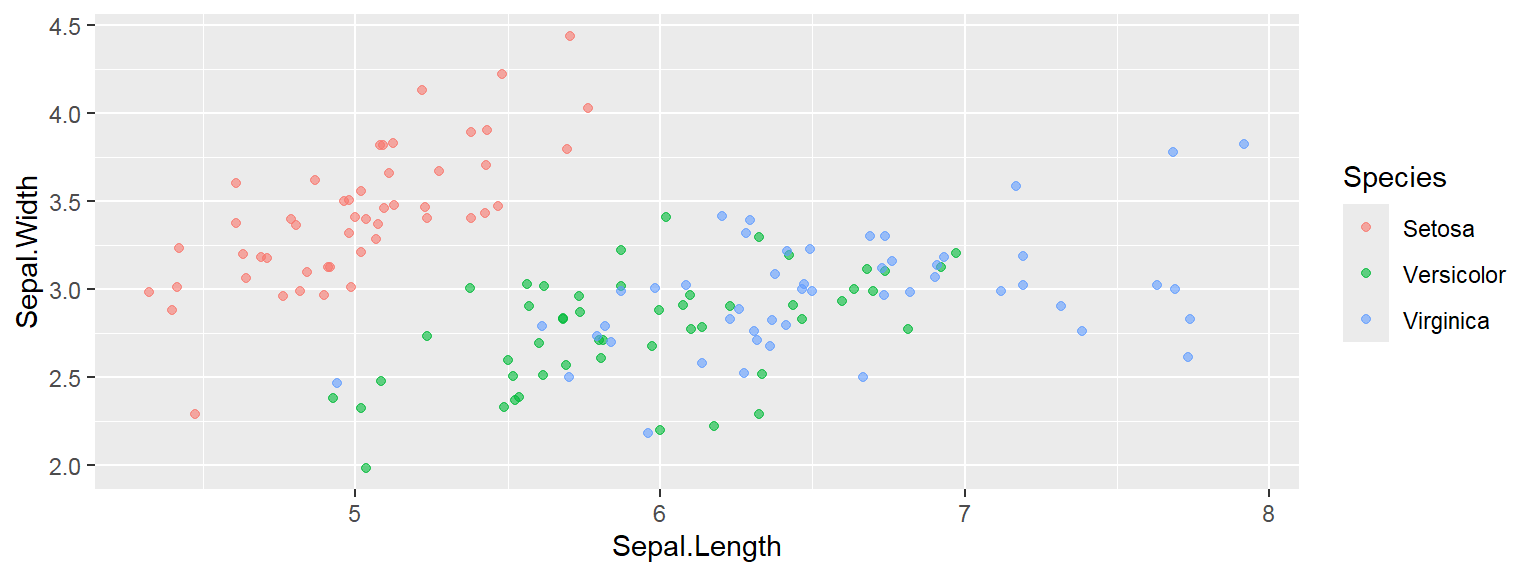
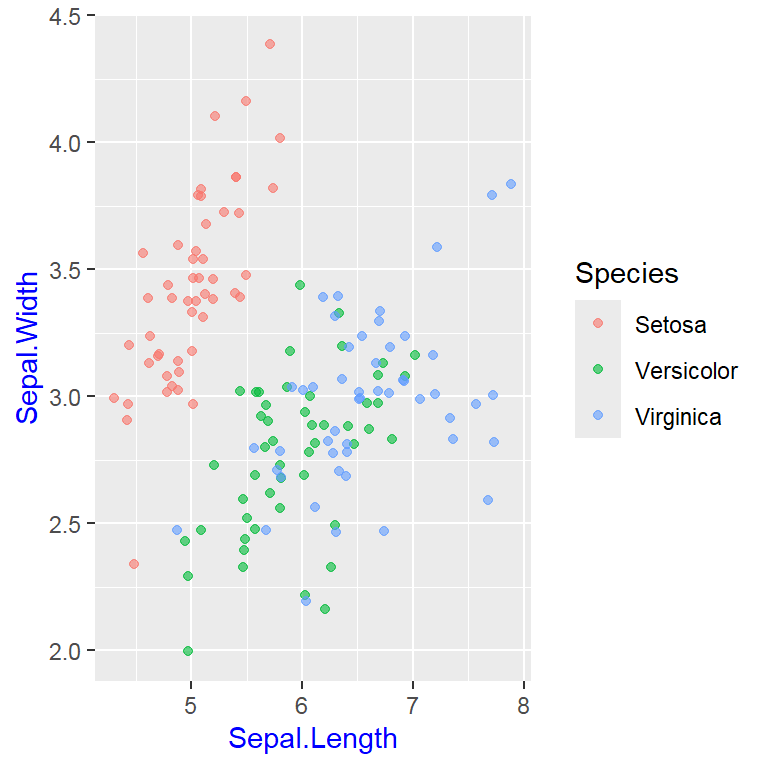
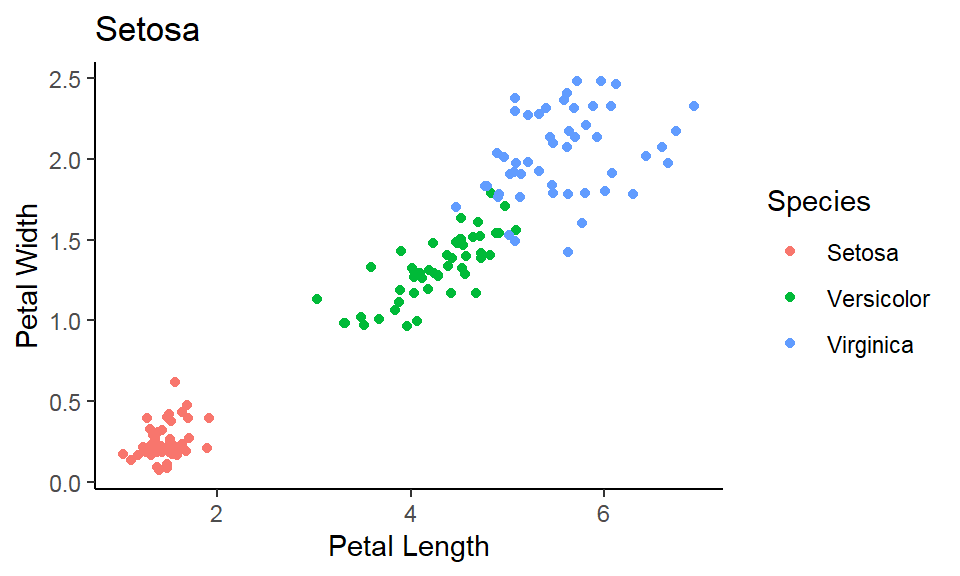
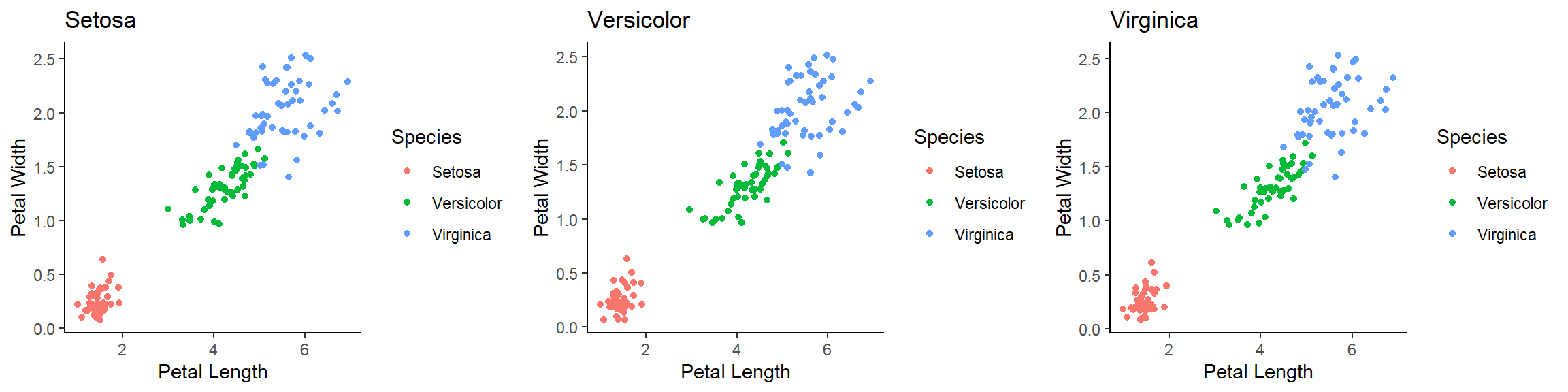
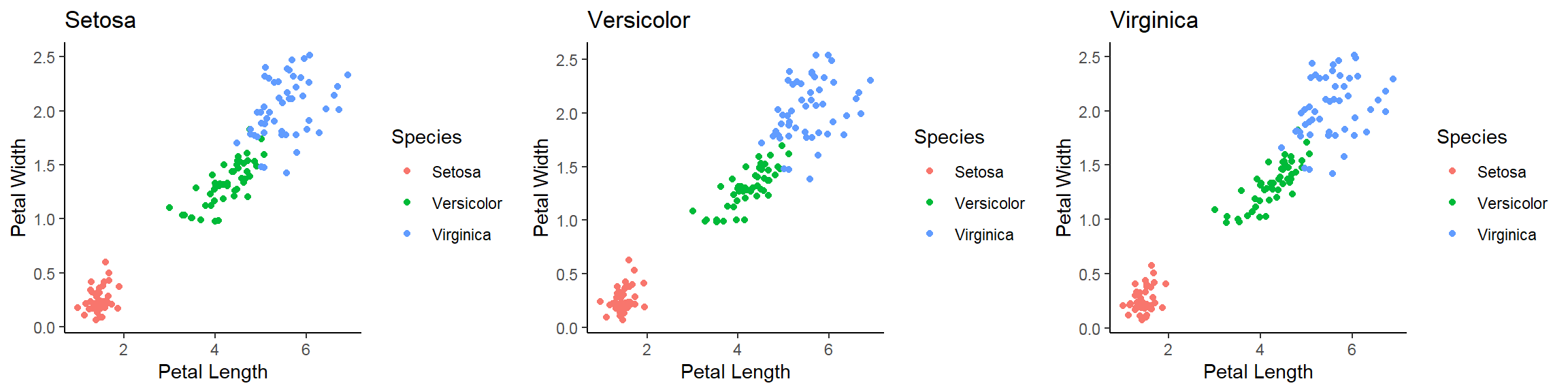
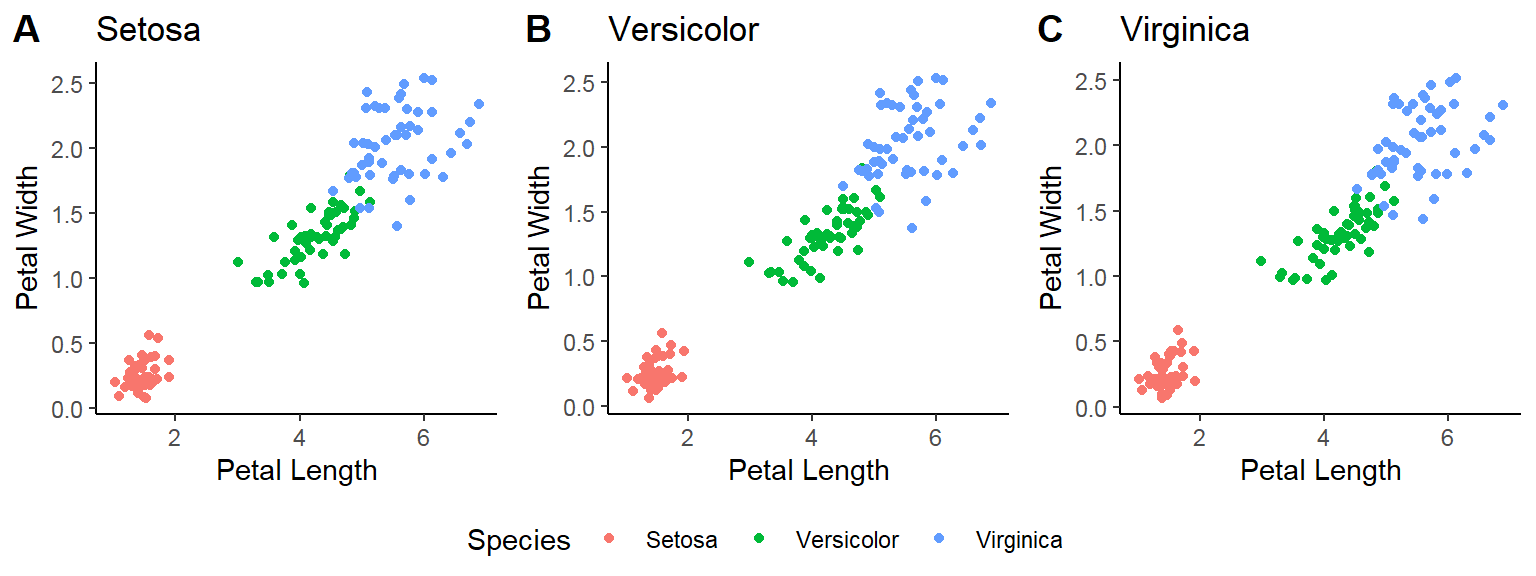
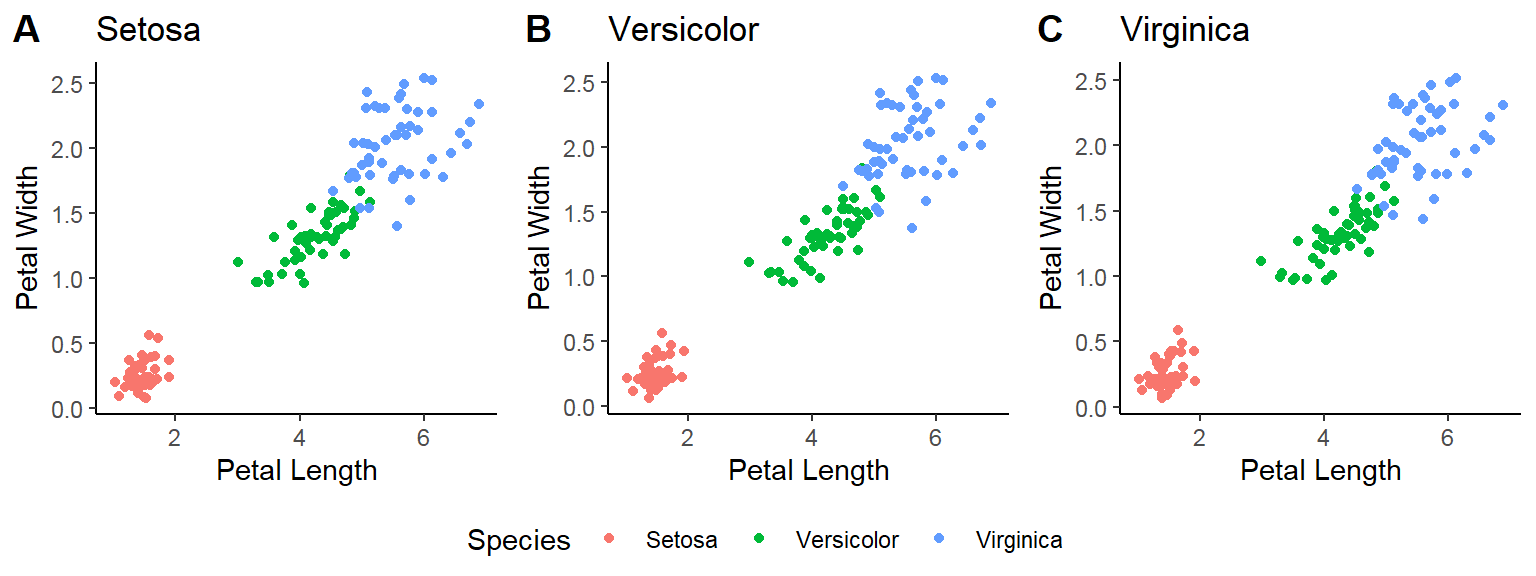
3.2.1 Main content
High-dimensional data
- Feature projection / Manifold learning
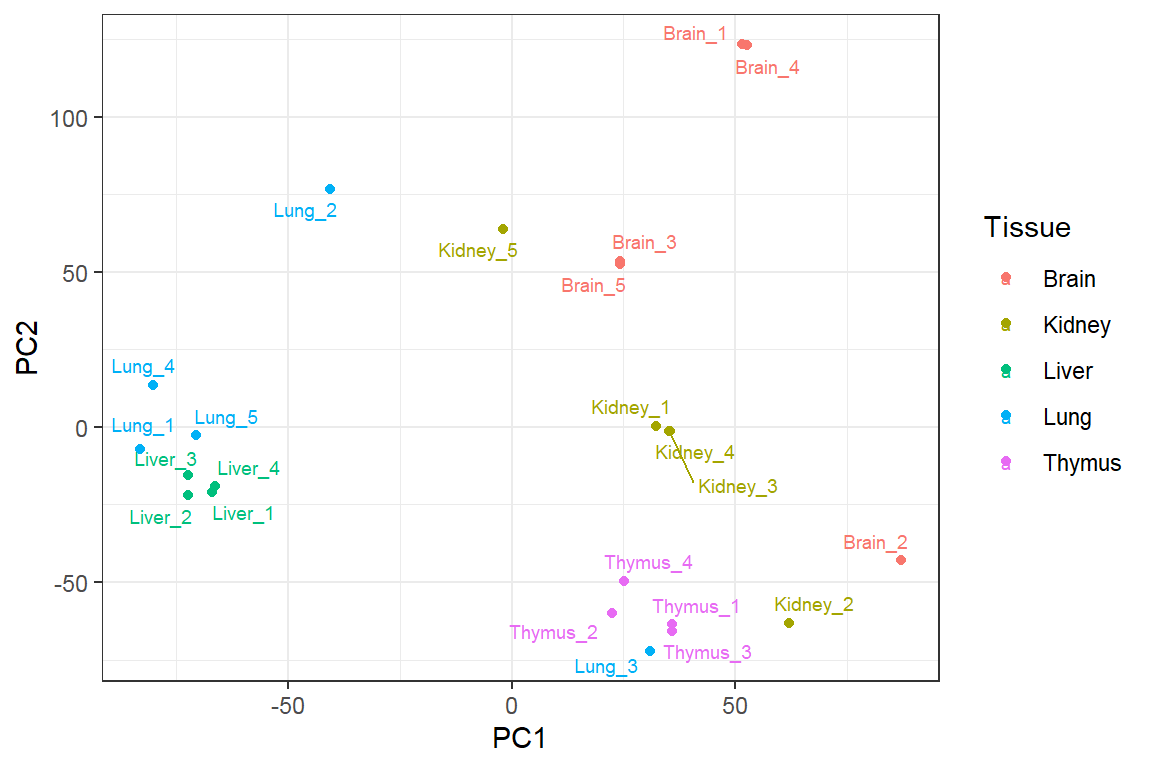
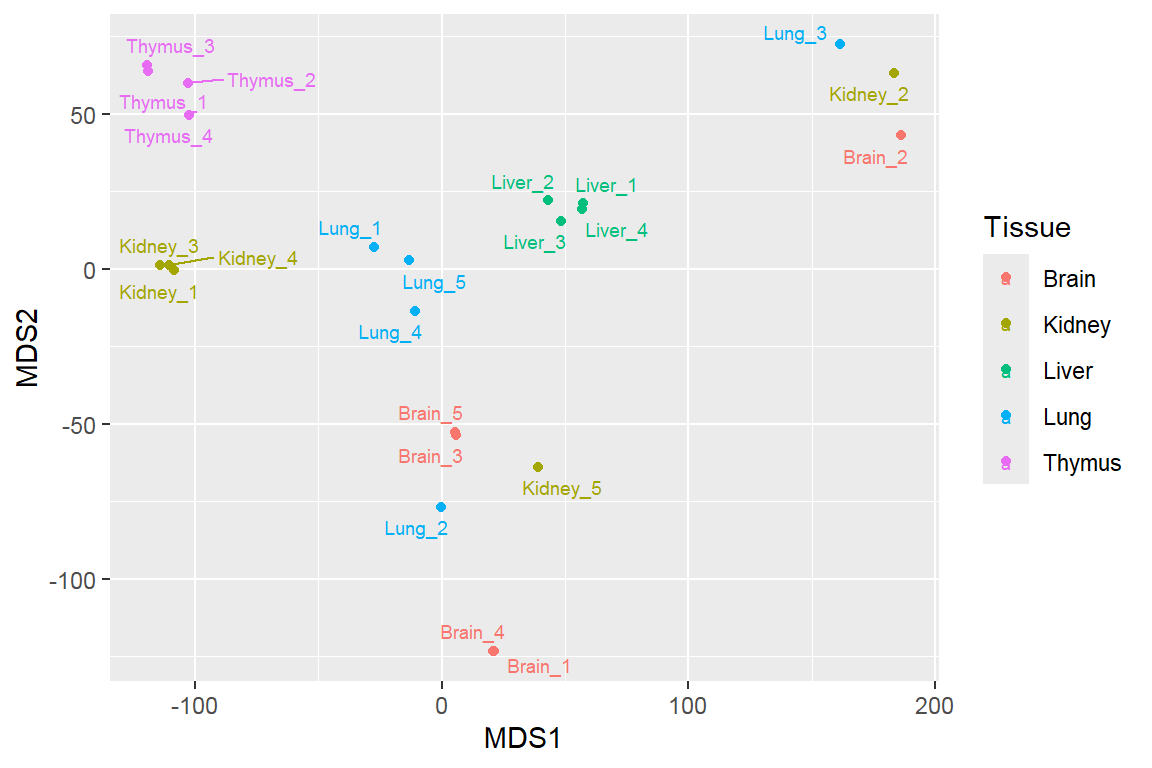
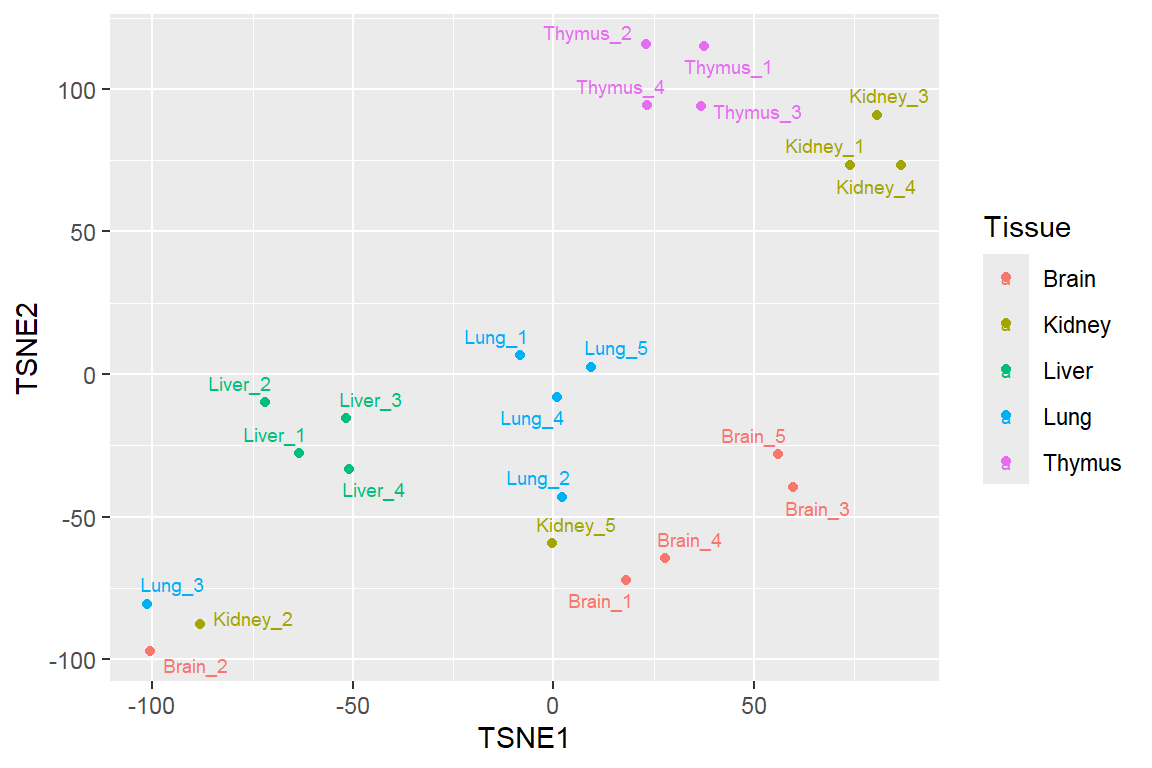
- 4 popular feature projection techniques: PCA, MDS, t-SNE, UMAP
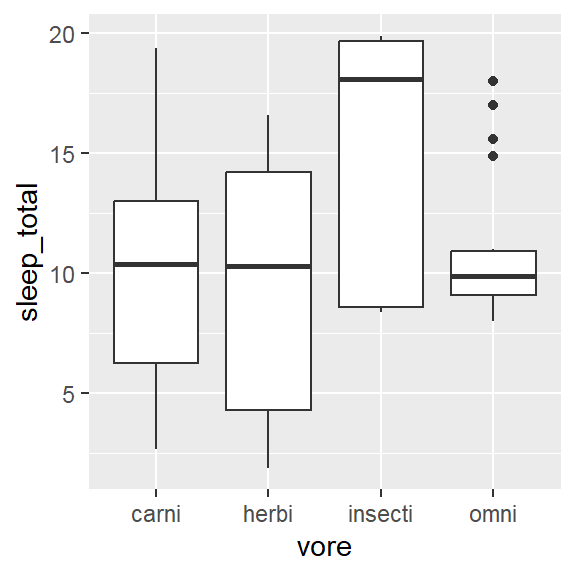
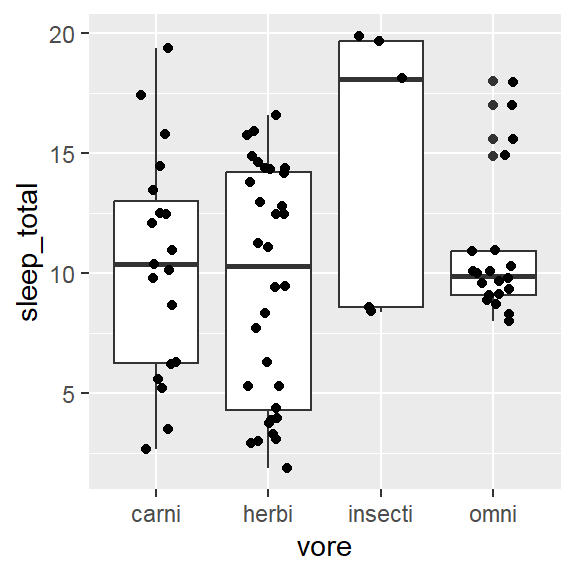
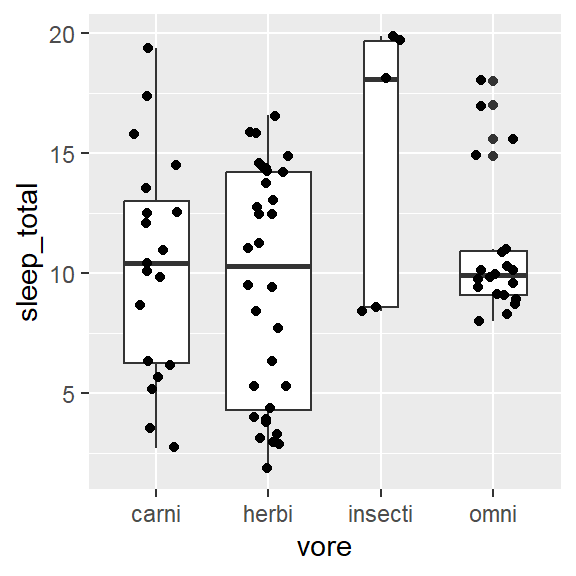
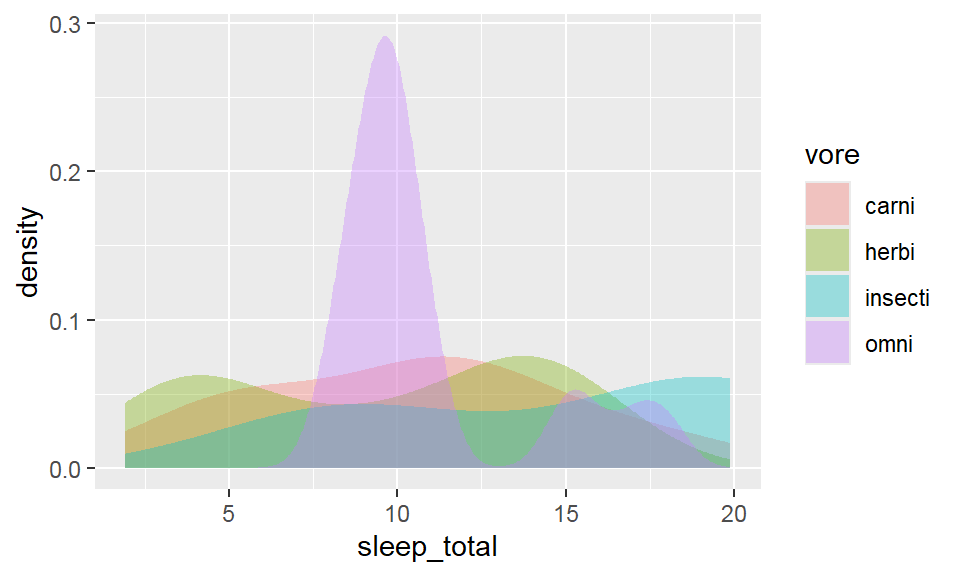
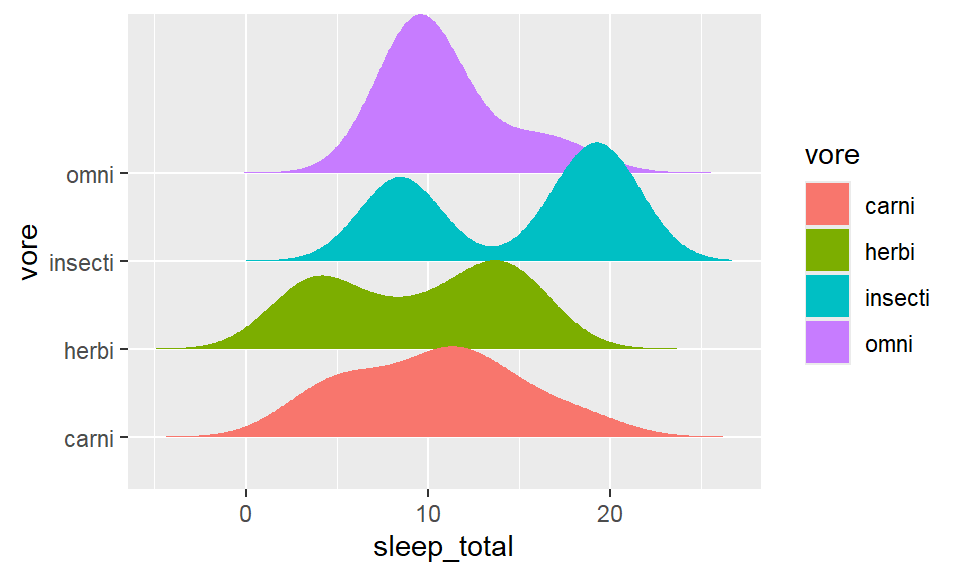
Distribution plot
- Within 1 variable:
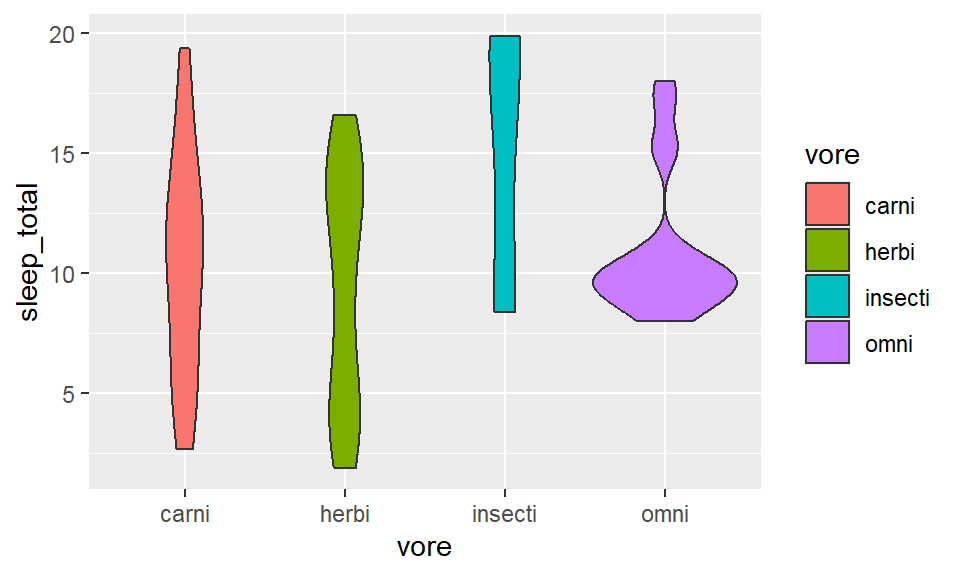
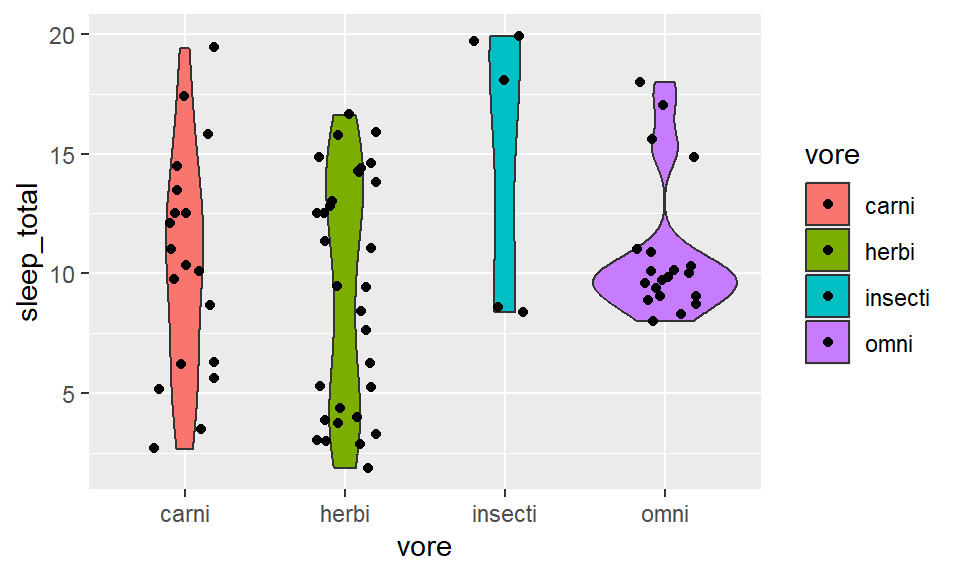
- Weighted box/ violin
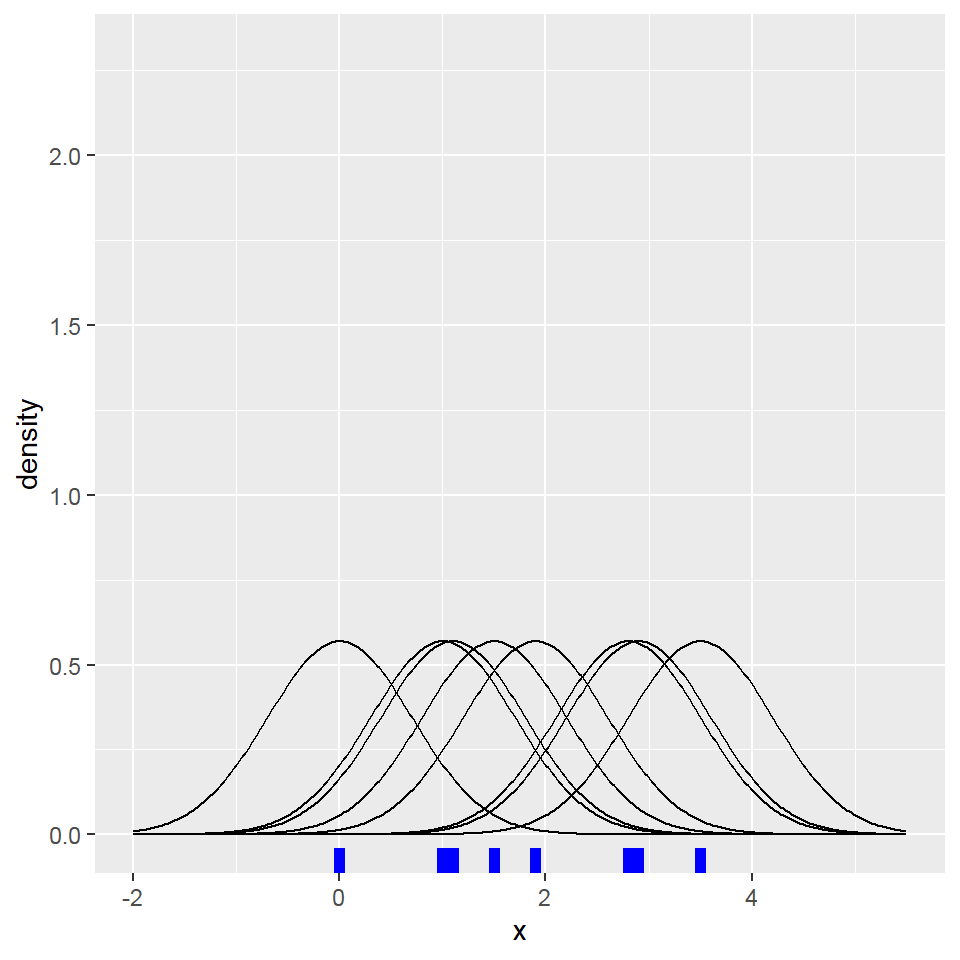
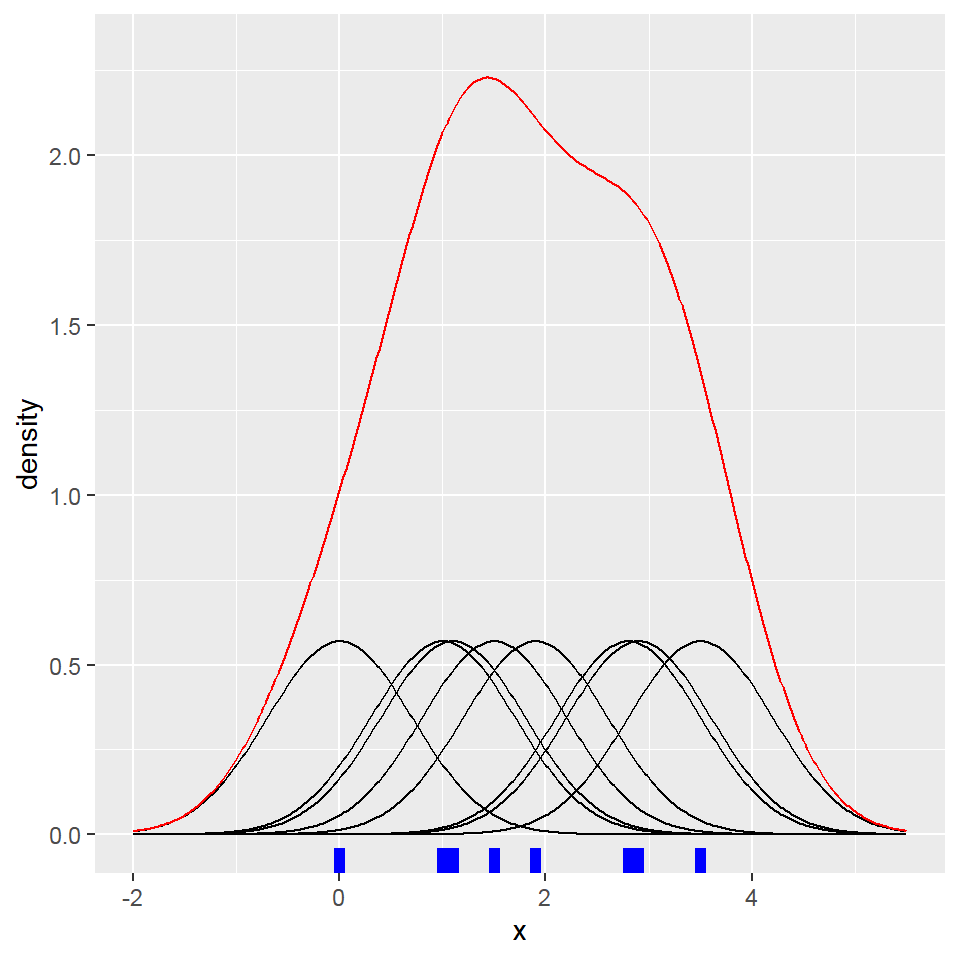
- Density
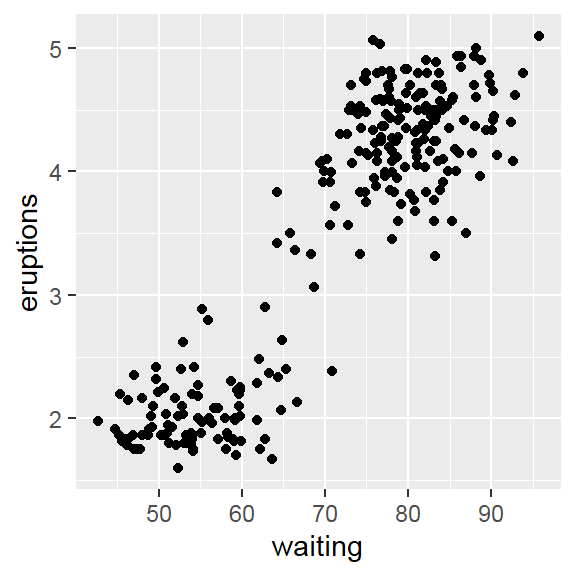
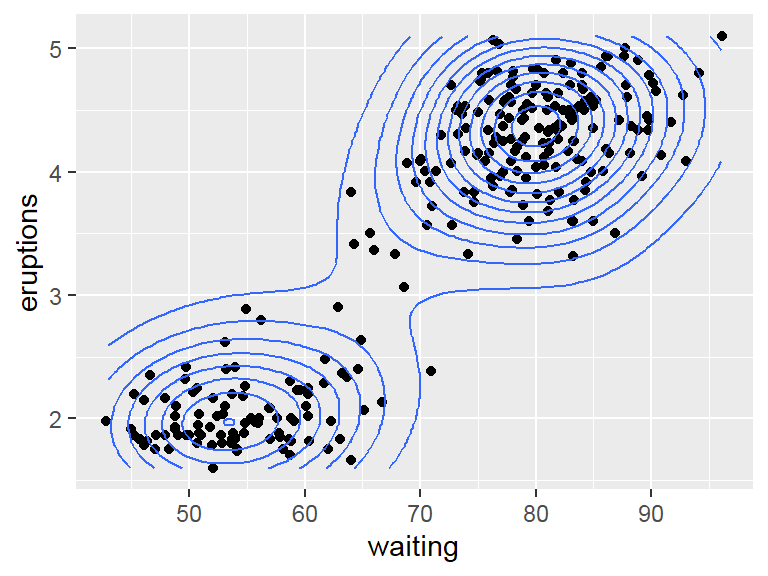
- 2 Separate variables:
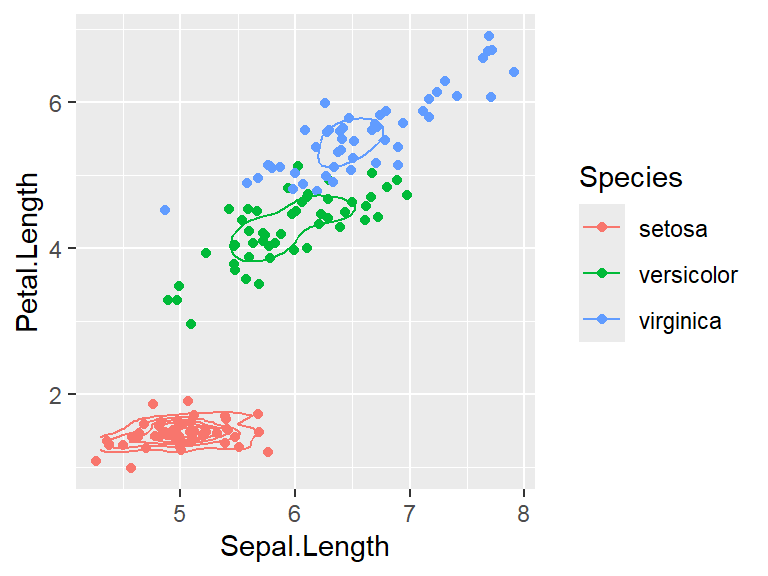
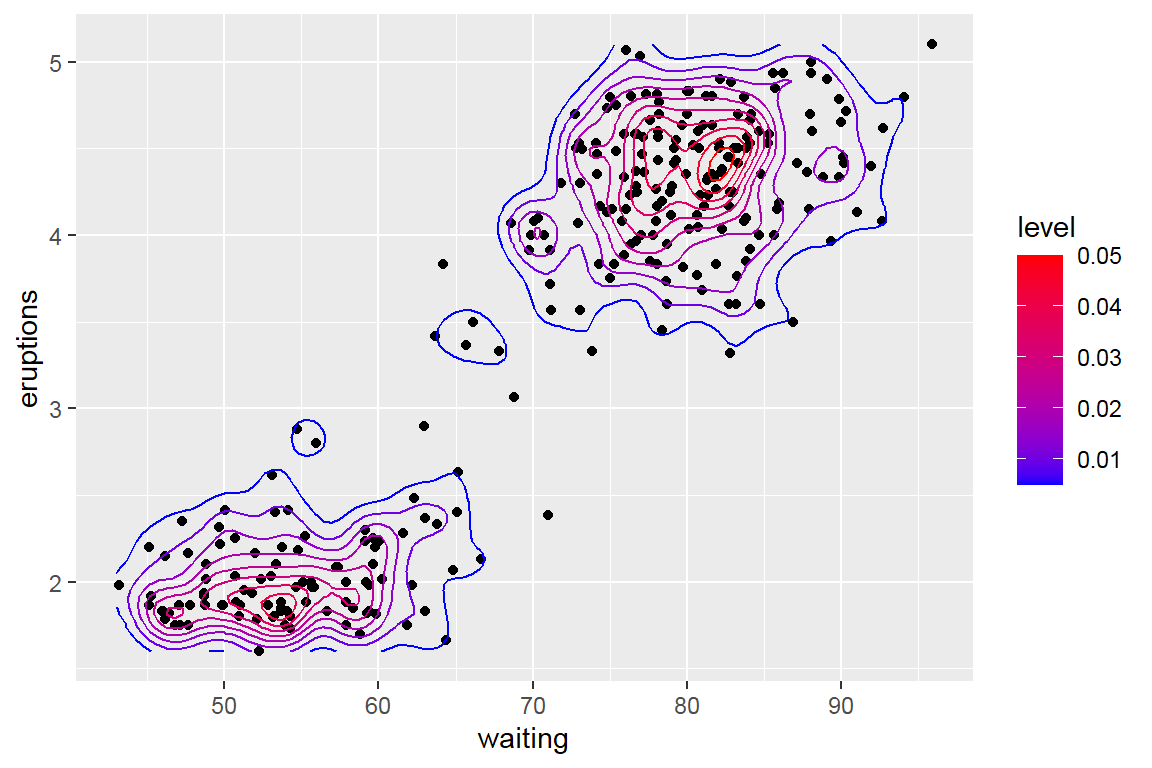
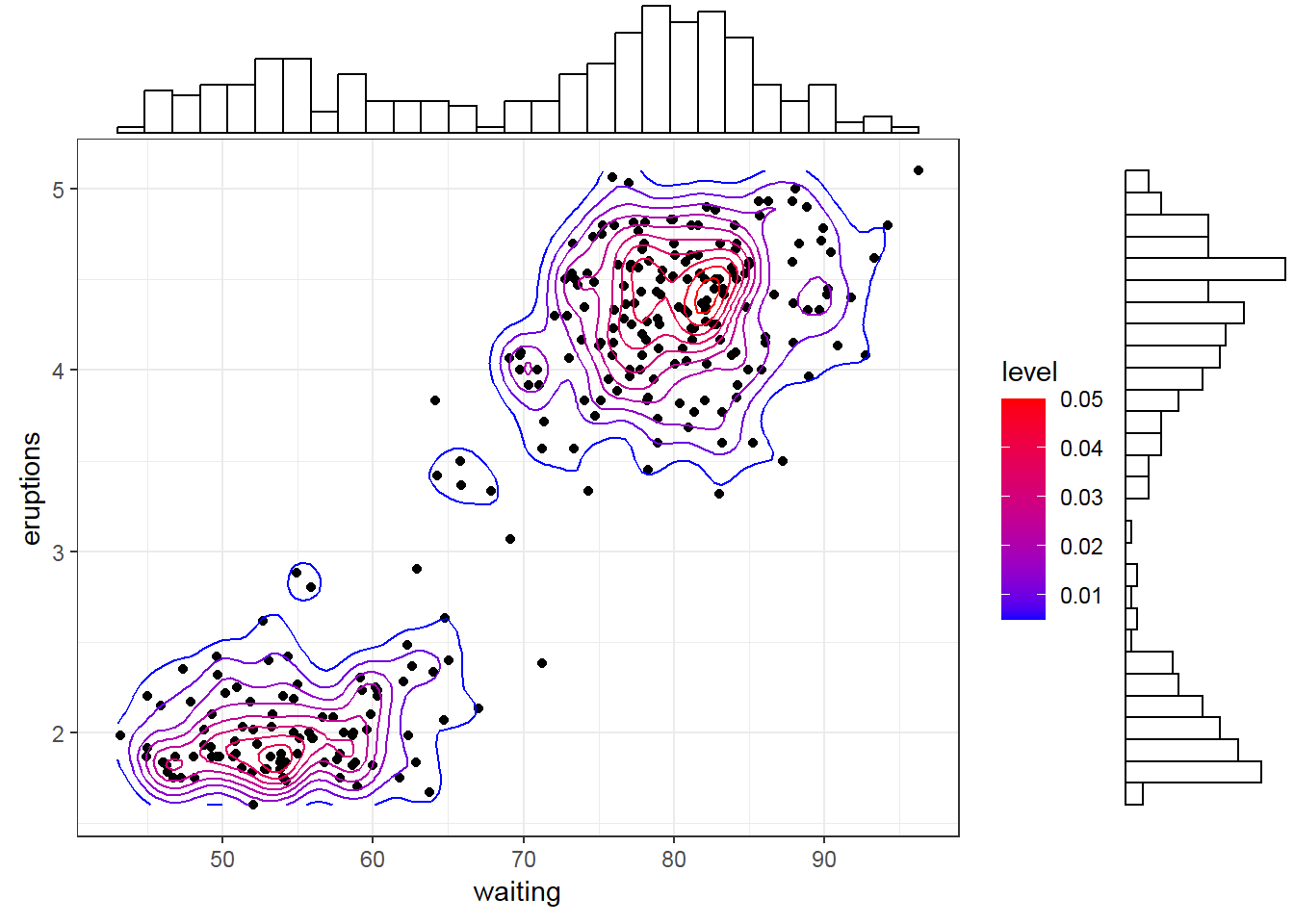
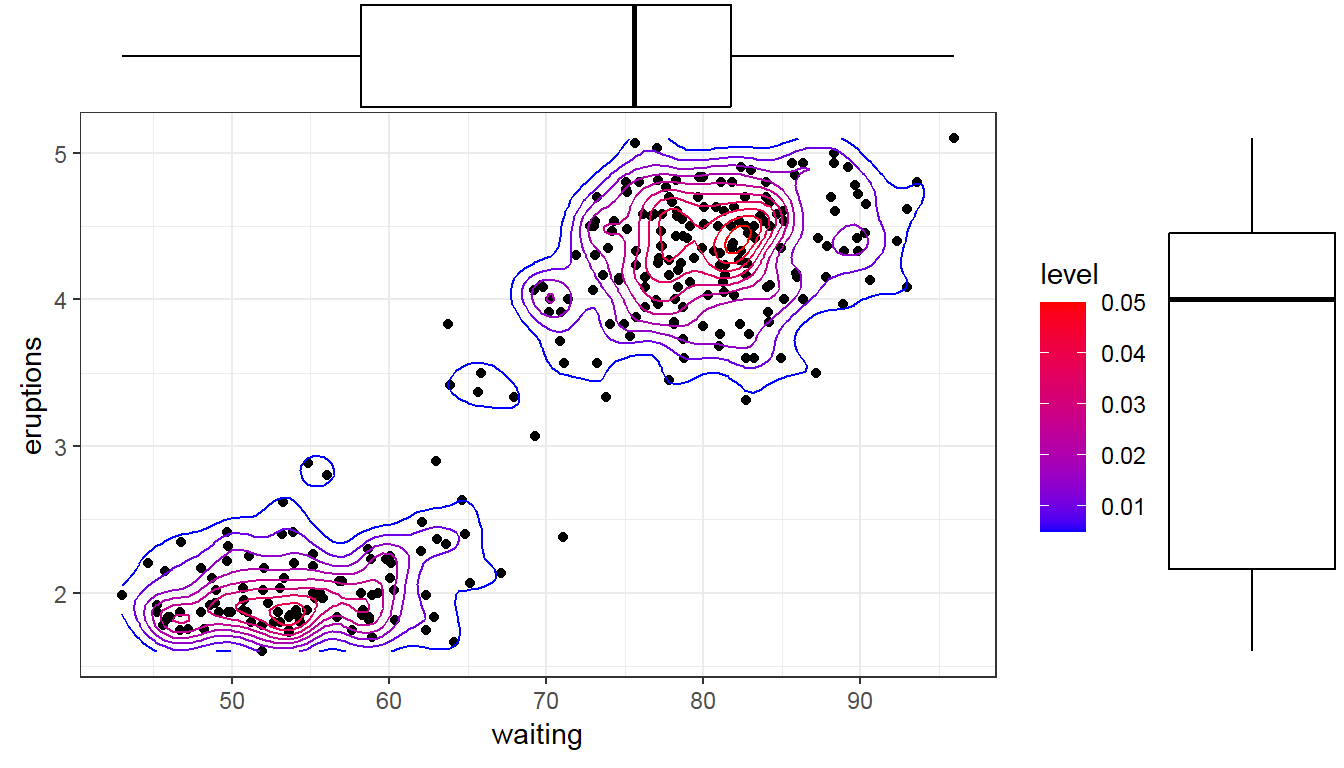
- 2D density
- Marginal histogram/ box plot
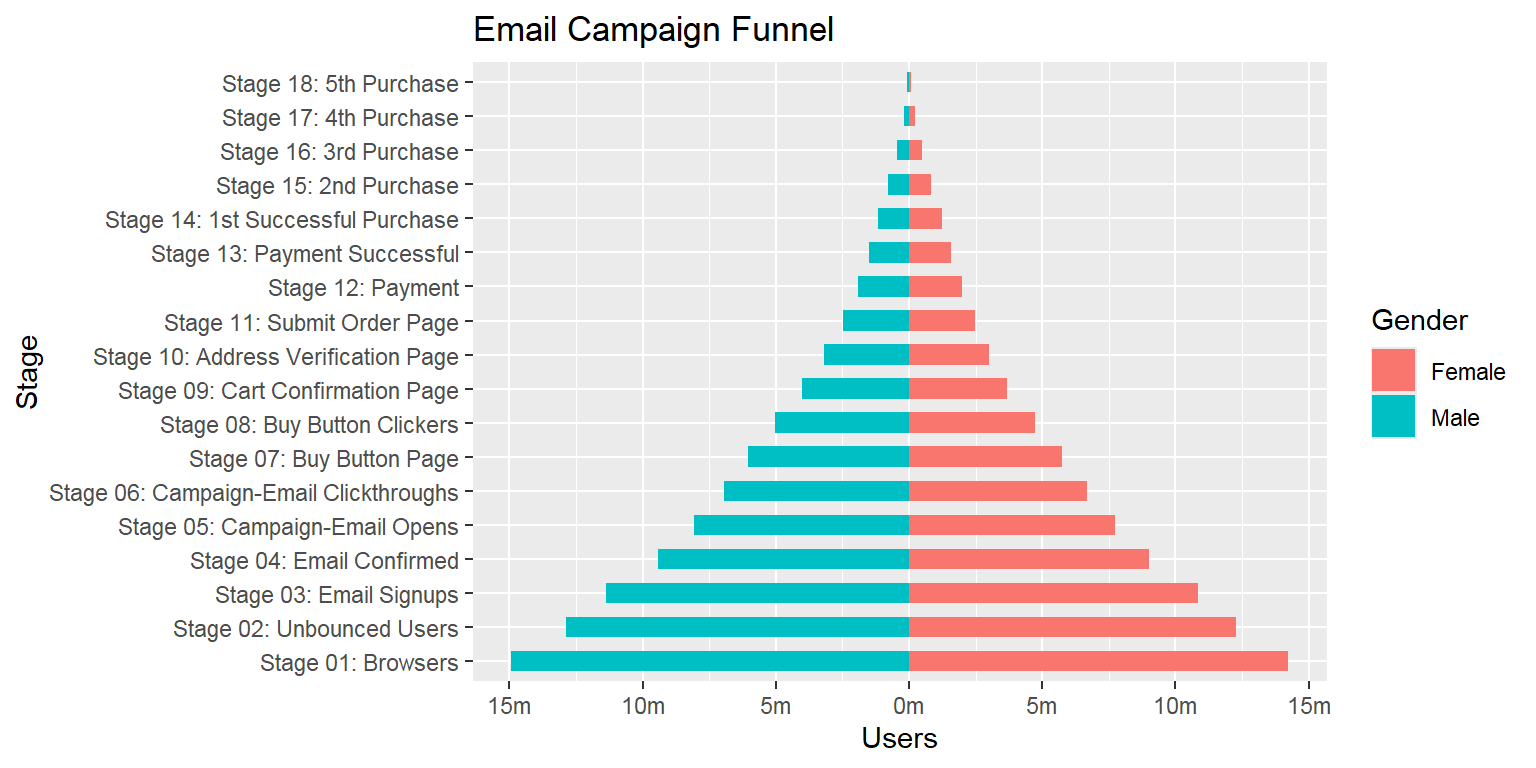
- Population pyramid
- Within 1 variable:
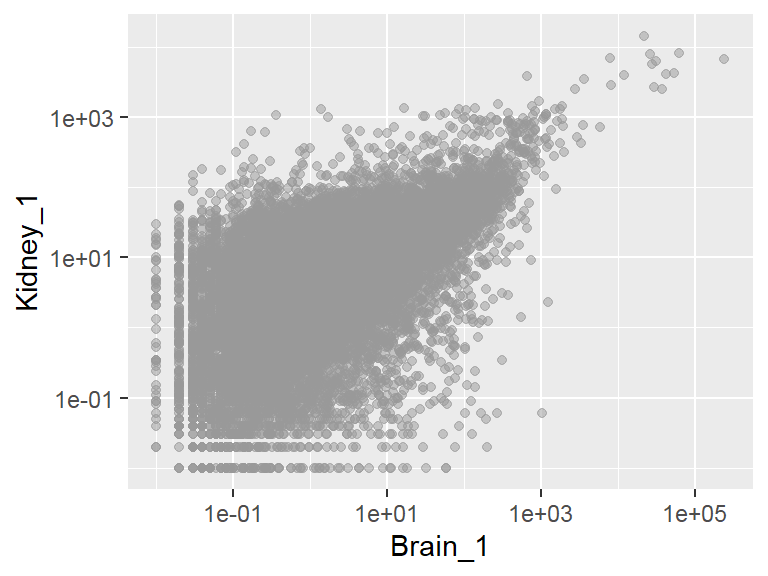
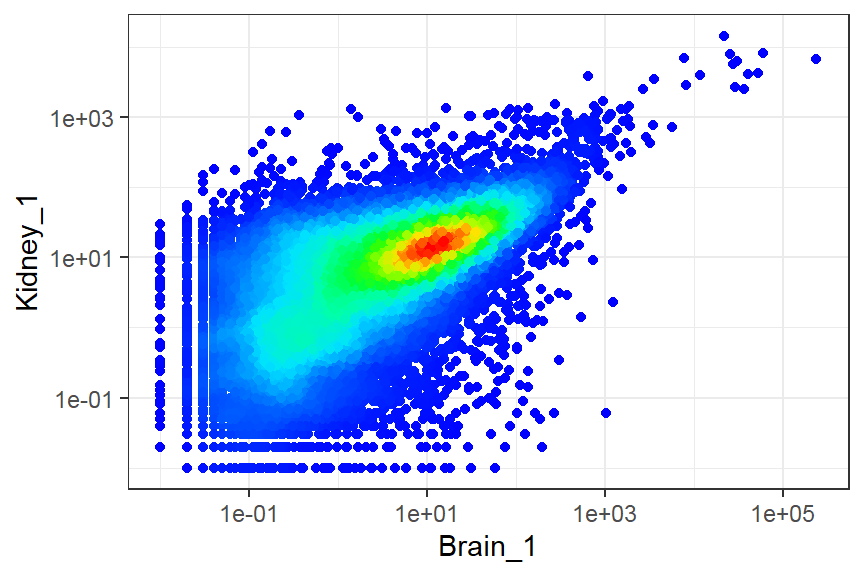
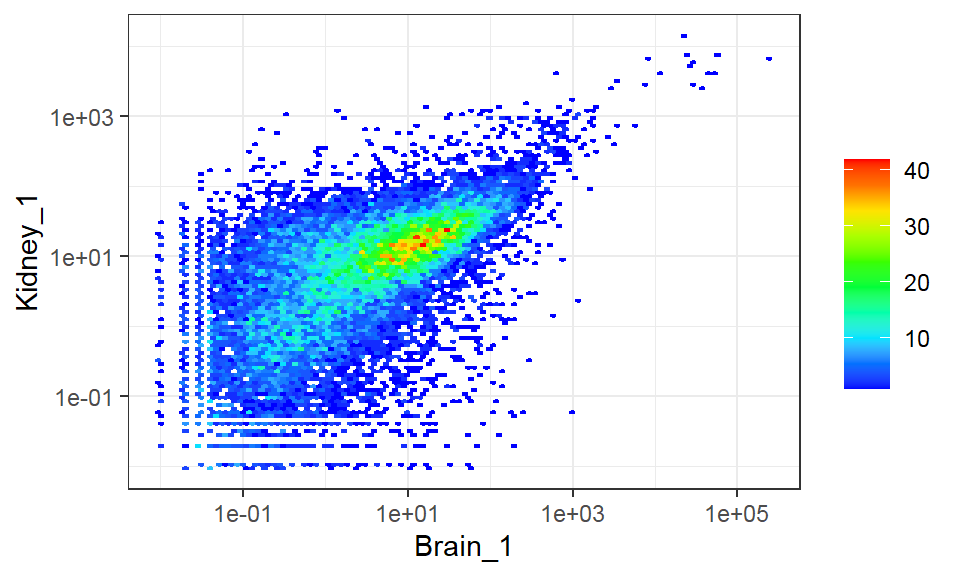
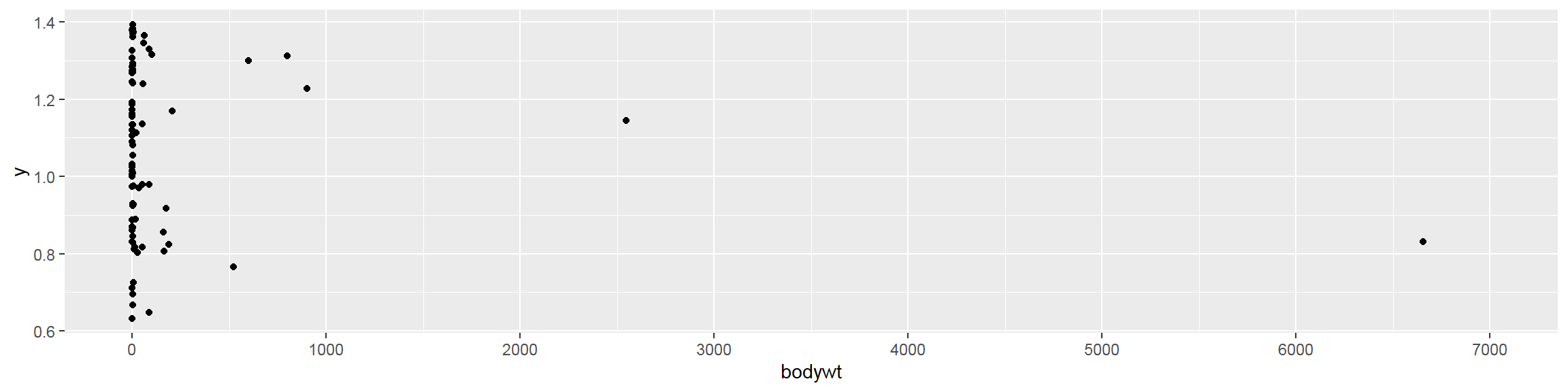
Deal with large number of observations
- Binned scatter
Deal with multi-dimensional data
- Feature projection/ Manifold learning >> high-dimensional
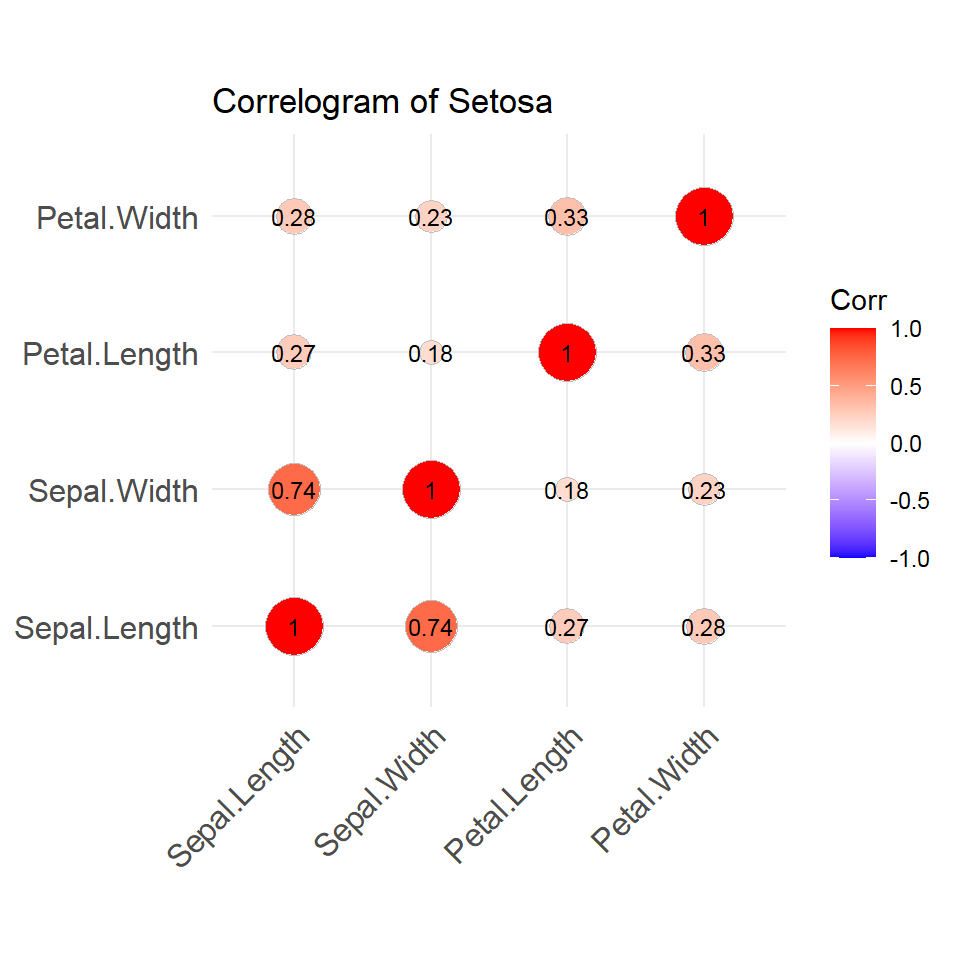
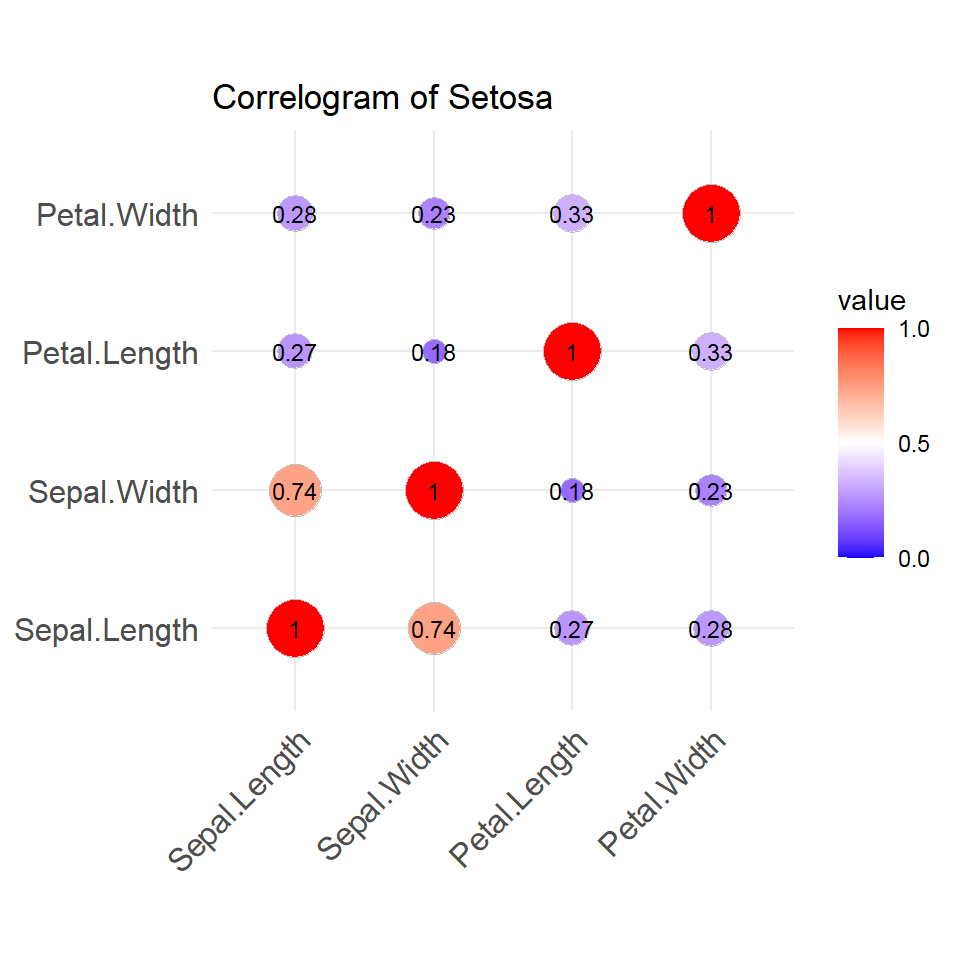
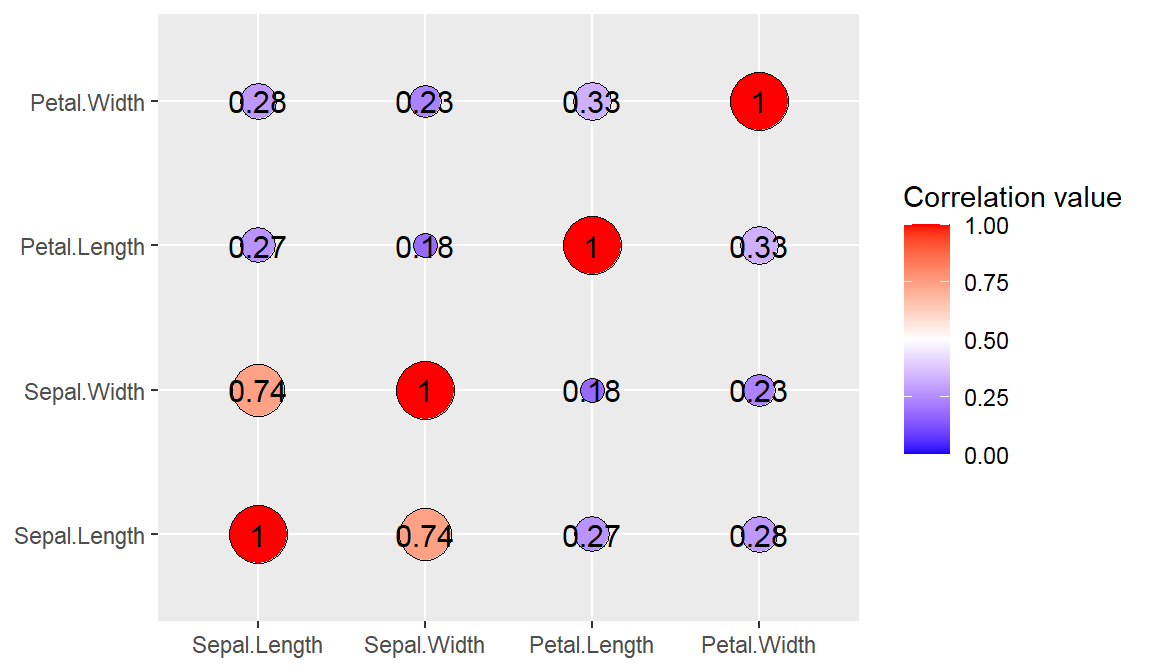
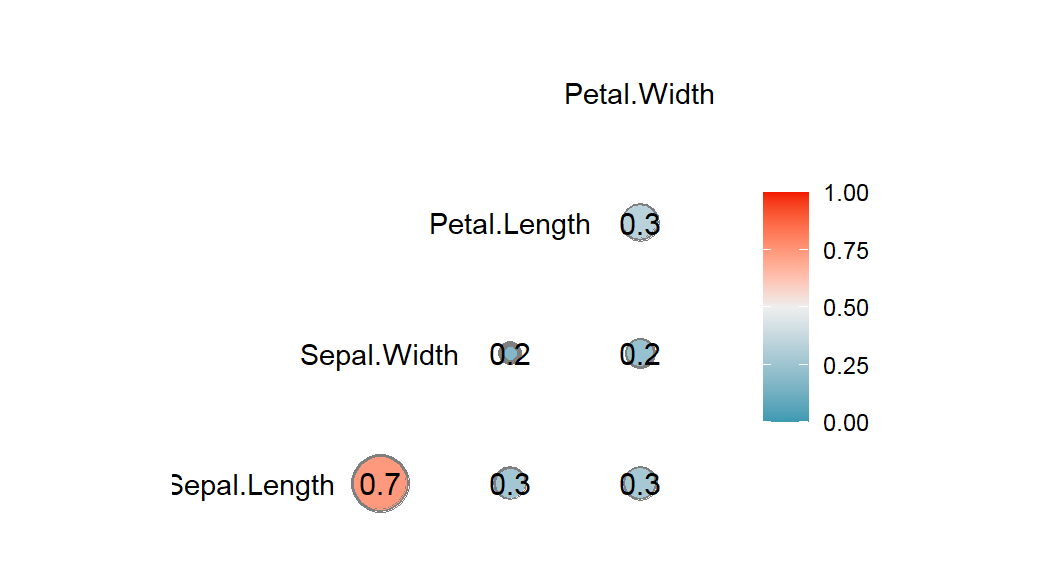
- Correlogram
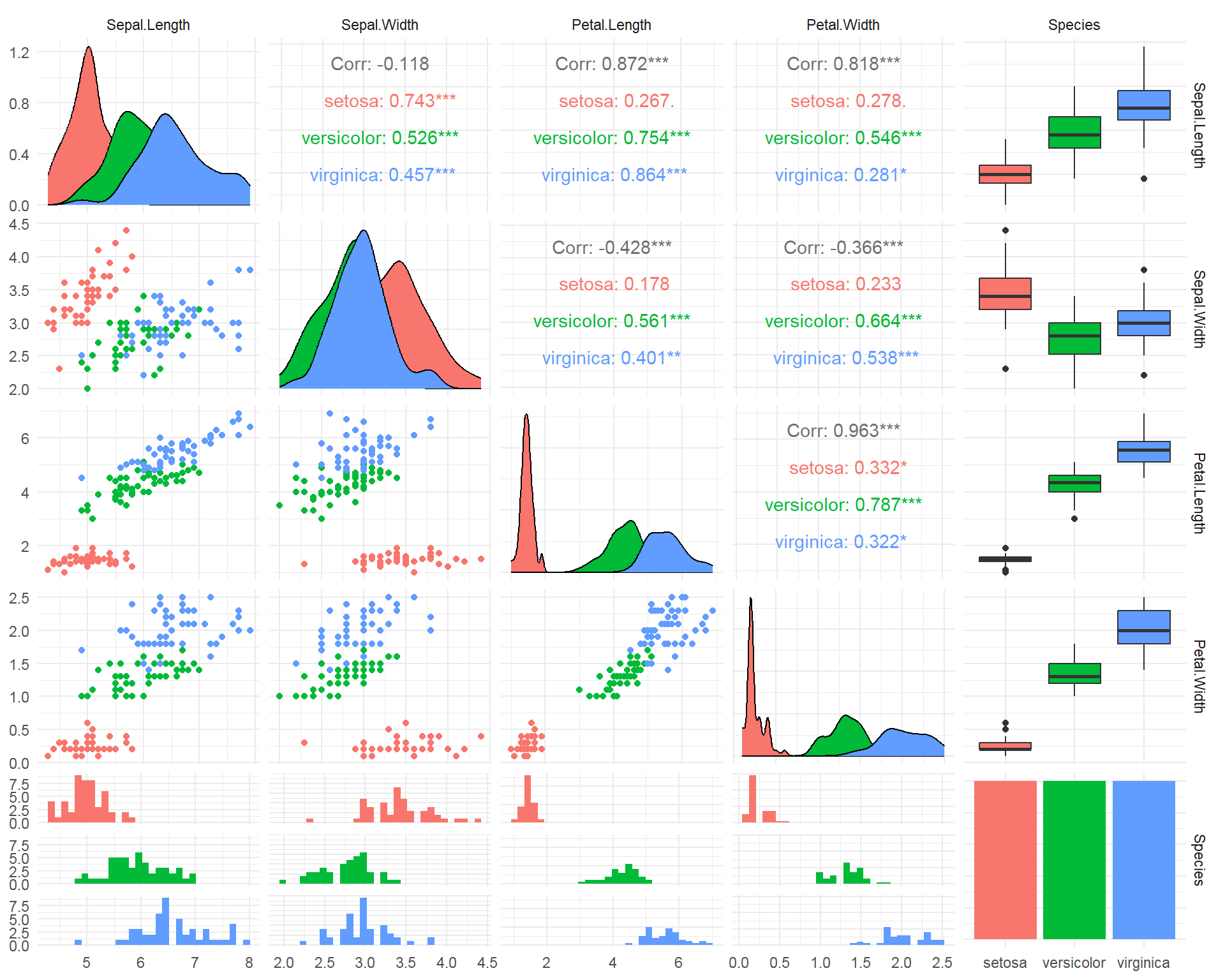
- SPLOM - Scatter PLOt Matrix
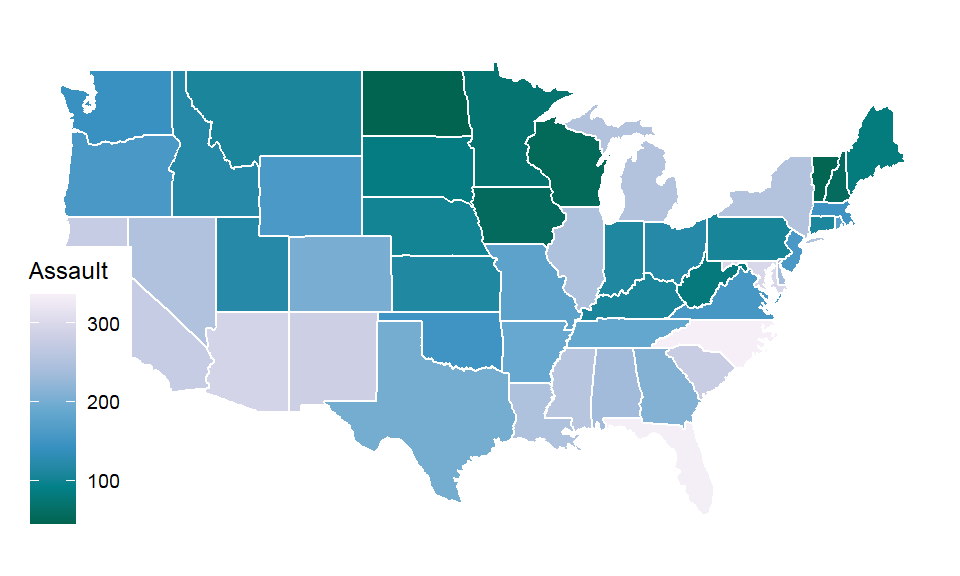
Map
- Chorophleth
Animation
Honorable mentions
plotly
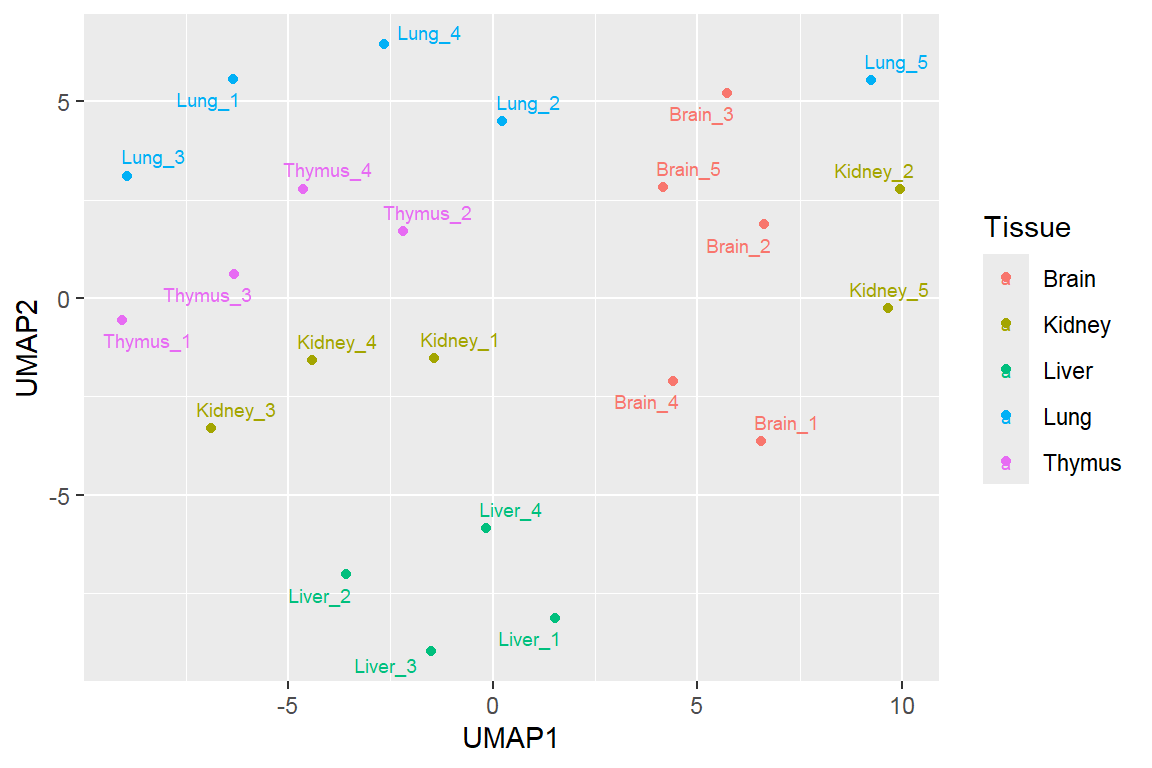
Feature projection/ Manifold learning
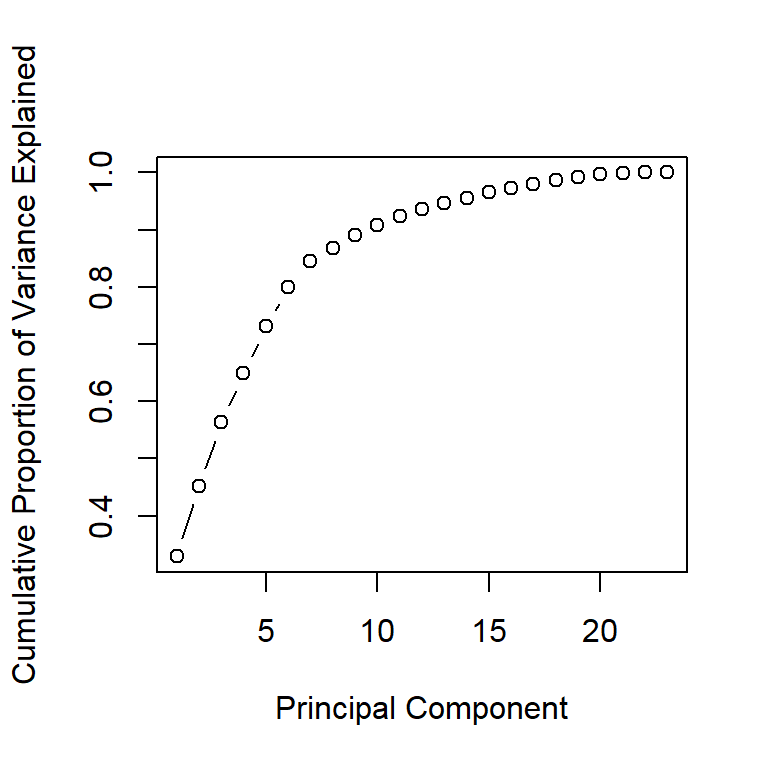
Principal Component Analysis
Multidimensional scaling
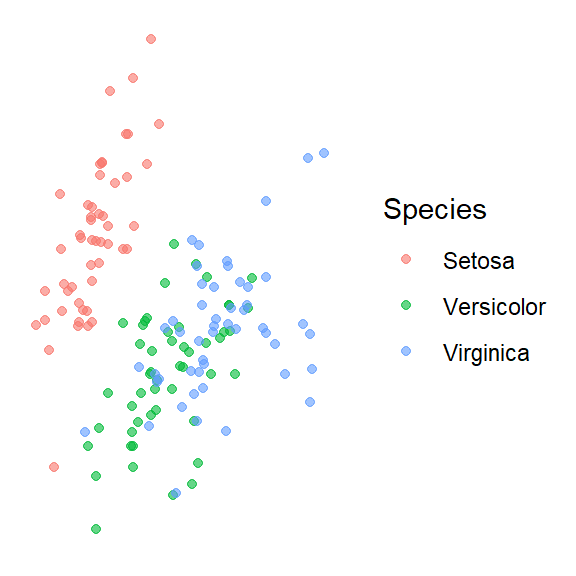
t-distributed stochastic neighbor embedding (t-SNE)
Uniform manifold approximation and projection (UMAP)
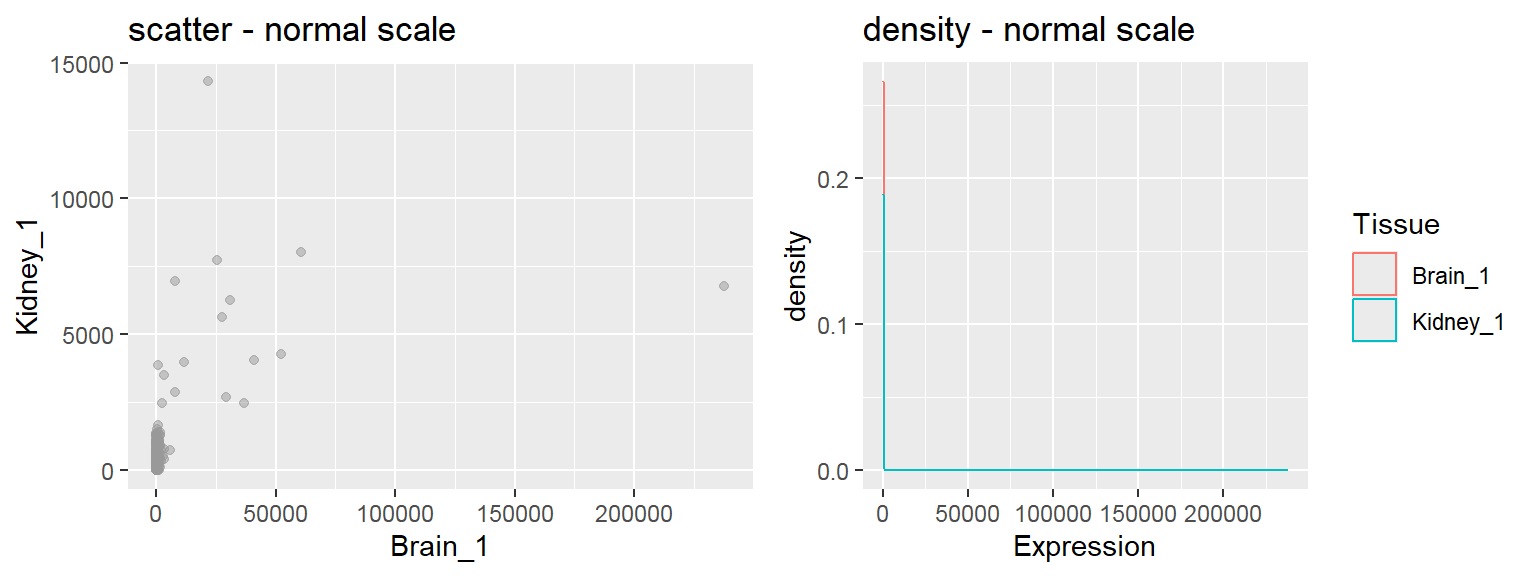
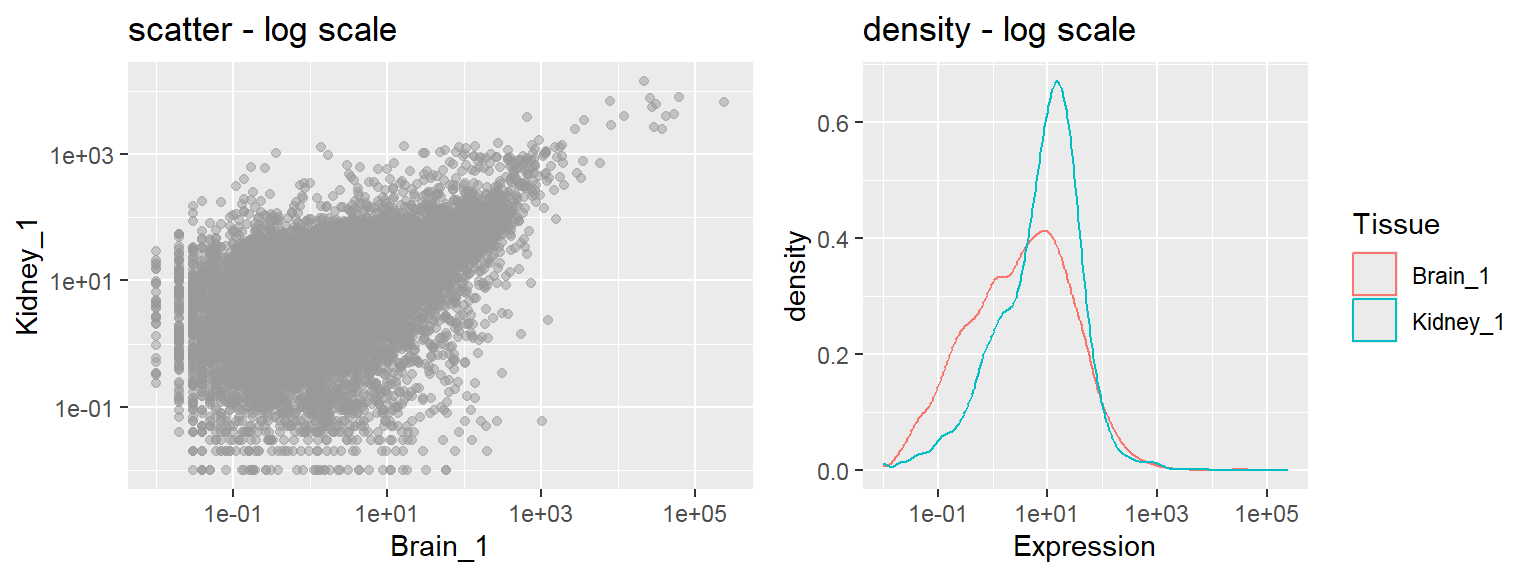
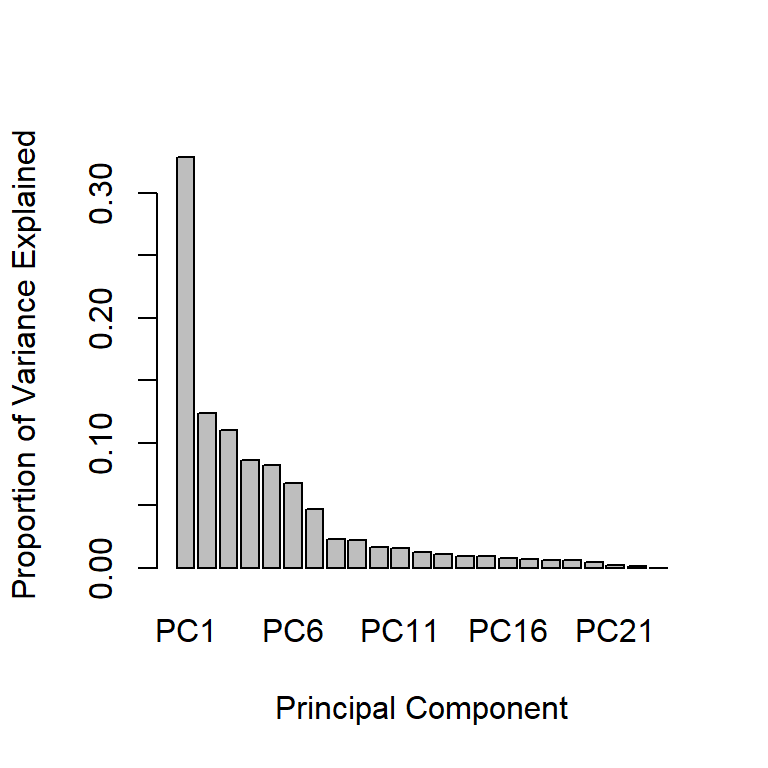 - In addition to the individual variance explained plots, also the cumulative variance explained is frequently looked at.
- In addition to the individual variance explained plots, also the cumulative variance explained is frequently looked at.