Chapter 9 Logical operators for matching
9.1 Logical operators to match elements
Oftentimes, we encounter different analysis tools that require multiple input datasets. It is not uncommon for these inputs to need to have the same row names, column names, or unique identifiers in the same order to perform the analysis. Therefore, knowing how to reorder datasets and determine whether the data matches is an important skill.
In our use case, we will be working with genomic data. We have gene expression data generated by RNA-seq, which we had downloaded previously; in addition, we have a metadata file corresponding to the RNA-seq samples. The metadata contains information about the samples present in the gene expression file, such as which sample group each sample belongs to and any batch or experimental variables present in the data.
Let’s read in our gene expression data (RPKM matrix) that we downloaded previously:
Take a look at the first few lines of the data matrix to see what’s in there.
It looks as if the sample names (header) in our data matrix are similar to the row names of our metadata file, but it’s hard to tell since they are not in the same order. We can do a quick check of the number of columns in the count data and the rows in the metadata and at least see if the numbers match up.
What we want to know is, do we have data for every sample that we have metadata?
9.2 The %in% operator
This operator is well-used and convenient once you get the hang of it. The operator is known as exactly in and is used with the following syntax:
It will take each element from vector1 as input, one at a time, and evaluate if the element is present in vector2. The two vectors do not have to be the same size. This operation will return a vector containing logical values to indicate whether or not there is a match. The new vector will be of the same length as vector1. Take a look at the example below:
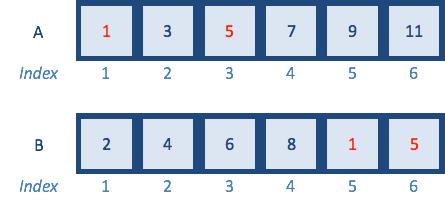
A <- c(1,3,5,7,9,11) # odd numbers
B <- c(2,4,6,8,10,12) # even numbers
# test to see if each of the elements of A is in B
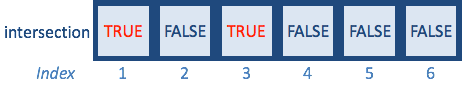
A %in% BSince vector A contains only odd numbers and vector B contains only even numbers, the operation returns a logical vector containing six FALSE, suggesting that no element in vector A is present in vector B. Let’s change a couple of numbers inside vector B to match vector A:
The returned logical vector denotes which elements in A are also in B - the first and third elements, which are 1 and 5.
Note: this function is not reversible; i.e. B %in% A will give a different answer.
We saw previously that we could use the output from a logical expression to subset data by returning only the values corresponding to TRUE. Therefore, we can use the output logical vector to subset our data, and return only those elements in A, which are also in B by returning only the TRUE values:

In these previous examples, the vectors were so small that it’s easy to check every logical value by eye; but this is not practical when we work with large datasets (e.g. a vector with 1000 logical values). Instead, we can use any function. Given a logical vector, this function will tell you whether at least one value is TRUE. It provides us a quick way to assess if any of the values contained in vector A are also in vector B:
The all function is also useful. Given a logical vector, it will tell you whether all values are TRUE. If there is at least one FALSE value, the all function will return a FALSE. We can use this function to assess whether all elements from vector A are contained in vector B.
Using the
AandBvectors created above, evaluate each element inBto see if there is a match inASubset the
Bvector to only return those values that are also inA.
Suppose we had two vectors containing same values. How can we check if those values are in the same order in each vector? In this case, we can use == operator to compare each element of the same position from two vectors. The operator returns a logical vector indicating TRUE/FALSE at each position. Then we can use all() function to check if all values in the returned vector are TRUE. If all values are TRUE, we know that these two vectors are the same. Unlike %in% operator, == operator requires that two vectors are of equal length.
A <- c(10,20,30,40,50)
B <- c(50,40,30,20,10) # same numbers but backwards
# test to see if each element of A is in B
A %in% B
# test to see if each element of A is in the same position in B
A == B
# use all() to check if they are a perfect match
all(A == B)Let’s try this on our genomic data, and see whether we have metadata information for all samples in our expression data. We’ll start by creating two vectors: one is the rownames of the metadata, and one is the colnames of the RPKM data. These are base functions in R which allow you to extract the row and column names as a vector:
Now check to see that all of x are in y:
Note that we can use nested functions in place of x and y and still get the same result:
We know that all samples are present, but are they in the same order?
Looks like all of the samples are there, but they need to be reordered. To reorder our genomic samples, we will learn different ways to reorder data in our next lesson. But before that, let’s work on exercise 2 to consolidate concepts from this lesson.
We have a list of 6 marker genes that we are very interested in. Our goal is to extract count data for these genes using the %in% operator from the rpkm_data data frame, instead of scrolling through rpkm_data and finding them manually.
First, let’s create a vector called important_genes with the Ensembl IDs of the 6 genes we are interested in:
important_genes <- c("ENSMUSG00000083700", "ENSMUSG00000080990",
"ENSMUSG00000065619", "ENSMUSG00000047945", "ENSMUSG00000081010",
"ENSMUSG00000030970")Use the
%in%operator to determine if all of these genes are present in the row names of therpkm_datadata frame.Extract the rows from
rpkm_datathat correspond to these 6 genes using[]and the%in%operator. Double check the row names to ensure that you are extracting the correct rows.Bonus question: Extract the rows from
rpkm_datathat correspond to these 6 genes using[], but without using the%in%operator.