Chapter 8 Dataframes and matrices
8.1 Dataframes
Dataframes (and matrices) have 2 dimensions (rows and columns), so if we want to select some specific data from it we need to specify the “coordinates” we want from it. We use the same square bracket notation but rather than providing a single index, there are two indices required. Within the square bracket, row numbers come first followed by column numbers (and the two are separated by a comma). Let’s explore the metadata dataframe, shown below are the first six samples:
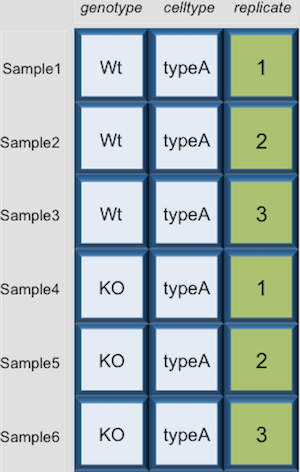
Let’s say we wanted to extract the wild type (Wt) value that is present in the first row and the first column. To extract it, just like with vectors, we give the name of the data frame that we want to extract from, followed by the square brackets. Now inside the square brackets we give the coordinates or indices for the rows in which the value(s) are present, followed by a comma, then the coordinates or indices for the columns in which the value(s) are present. We know the wild type value is in the first row if we count from the top, so we put a one, then a comma. The wild type value is also in the first column, counting from left to right, so we put a one in the columns space too.
Now let’s extract the value 1 from the first row and third column.
Now if you only wanted to select based on rows, you would provide the index for the rows and leave the columns index blank. The key here is to include the comma, to let R know that you are accessing a 2-dimensional data structure:
What kind of data structure does the output appear to be? We see that it is two-dimensional with row names and column names, so we can surmise that it’s likely a data frame.
If you were selecting specific columns from the data frame - the rows are left blank:
What kind of data structure does this output appear to be? It looks different from the data frame, and we really just see a series of values output, indicating a vector data structure. This happens by default. R automatically drops to the simplest data structure possible. Oftentimes however, we would like to keep our single column as a data frame. To do this, there is an argument we can add when subsetting called drop, by changing it’s value to FALSE the output is kept as a data frame.
Just like with vectors, you can select multiple rows and columns at a time. Within the square brackets, you need to provide a vector of the desired values.
We can extract consecutive rows or columns using the colon (:) to create the vector of indices to extract.
Alternatively, we can use the combine function (c()) to extract any number of rows or columns. Let’s extract the first, third, and sixth rows.
For larger datasets, it can be tricky to remember the column number that corresponds to a particular variable. It’s, therefore, often better to use column/row names to refer to extract particular values, and it makes your code easier to read and your intentions clearer.
# Extract the celltype column for the first three samples
metadata[c("sample1", "sample2", "sample3") , "celltype"] If you need to remind yourself of the column/row names, the following functions are helpful:
# Check column names of metadata data frame
colnames(metadata)
# Check row names of metadata data frame
rownames(metadata)If only a single column is to be extracted from a data frame, there is a useful shortcut available. If you type the name of the data frame, then the $, you have the option to choose which column to extract. For instance, let’s extract the entire genotype column from our dataset:
The output will always be a vector, and if desired, you can continue to treat it as a vector. For example, if we wanted the genotype information for the first five samples in metadata, we can use the square brackets ([]) with the indices for the values from the vector to extract:
Unfortunately, there is no equivalent $ syntax to select a row by name.
Exercises
- Return a data frame with only the
genotypeandreplicatecolumn values forsample2andsample8. - Return the fourth and ninth values of the
replicatecolumn. - Extract the
replicatecolumn as a data frame.
8.1.1 Selecting using indices with logical operators
With data frames, similar to vectors, we can use logical expressions to extract the rows or columns in the data frame with specific values. First, we need to determine the indices in a rows or columns where a logical expression is TRUE, then we can extract those rows or columns from the data frame.
For example, if we want to return only those rows of the data frame with the celltype column having a value of typeA, we would perform two steps:
Identify which rows in the celltype column have a value of
typeA.Use those TRUE values to extract those rows from the data frame.
To do this we would extract the column of interest as a vector, with the first value corresponding to the first row, the second value corresponding to the second row, so on and so forth. We use that vector in the logical expression. Here we are looking for values to be equal to typeA, so our logical expression would be:
This will output TRUE and FALSE values for the values in the vector. The first six values are TRUE, while the last six are FALSE. This means the first six rows of our metadata have a vale of typeA while the last six do not. We can save these values to a variable, which we can call whatever we would like; let’s call it logical_idx.
Now we can use those TRUE and FALSE values to extract the rows that correspond to the TRUE values from the metadata data frame. We will extract as we normally would a data frame with metadata[ , ], and we need to make sure we put the logical_idx in the row’s space, since those TRUE and FALSE values correspond to the ROWS for which the expression is TRUE/FALSE. We will leave the column’s space blank to return all columns.
8.1.2 Logical operators using the which() function
As you might have guessed, we can also use the which() function to return the indices for which the logical expression is TRUE. For example, we can find the indices where the celltype is typeA within the metadata dataframe:
This returns the values one through six, indicating that the first 6 values or rows are true, or equal to typeA. We can save our indices for which rows the logical expression is true to a variable we’ll call idx, but, again, you could call it anything you want.
Then, we can use these indices to indicate the rows that we would like to return by extracting that data as we have previously, giving the idx as the rows that we would like to extract, while returning all columns:
Let’s try another subsetting. Extract the rows of the metadata data frame for only the replicates 2 and 3. First, let’s create the logical expression for the column of interest (replicate):
This should return the indices for the rows in the replicate column within metadata that have a value of 2 or 3. Now, we can save those indices to a variable and use that variable to extract those corresponding rows from the metadata table.
Alternatively, instead of doing this in two steps, we could use nesting to perform in a single step:
Either way works, so use the method that is most intuitive for you.
So far we haven’t stored as variables any of the extractions/subsettings that we have performed. Let’s save this output to a variable called sub_meta:
Exercise
Subset the metadata dataframe to return only the rows of data with a genotype of KO.