Capítulo 1 Lectura Automática de Datos
Un dato es una representación simbólica (numérica, alfabética, algorítmica, espacial, etc.) de un atributo, variable cuantitativa o variable cualitativa y que perteneciente a un conjunto de elementos (población).

Los datos ciertamente son una buena base para generar información de calidad y para la construcción de conocimiento. ¿Lo serán para la sabiduría?
A continuación, veremos diferentes tipos de datos y programas utilizados para su descarga y lectura automática en R.
1.1 Descarga de datos de internet
La función download.file() es útil para la descarga de datos de tablas de internet (p.e. txt, csv). Notar que el archivo se salva en el directorio de trabajo. Parámetros importantes son url, destfile, method. La funciónread.csv() se usa para leer los archivos en formato .csv. Algunos de los parámetros importantes son head (encabezado) y sep (tipo de separador), dec (para definir decimales) y row.names (para dar nombre a filas). Se utilizará de ejemplo base de datos de las Naciones Unidas sobre cantidad de especies amenazadas por país http://data.un.org/
URL <- "http://data.un.org/_Docs/SYB/CSV/SYB64_313_202110_Threatened%20Species.csv"
download.file(URL, destfile = "especies_amenazadas.csv")
file = read.csv("especies_amenazadas.csv", head = TRUE, sep =",")Ahora revisamos rápidamente la estructura de los datos con la función str() que permite ver el número de registros (filas) y la cantidad de variables (columnas). La función str() es probablemente una de las funciones más utilizadas en R
str(file)
#> 'data.frame': 6921 obs. of 7 variables:
#> $ T23 : chr "Region/Country/Area" "4" "4" "4" ...
#> $ Threatened.species: chr "" "Afghanistan" "Afghanistan" "Afghanistan" ...
#> $ X : chr "Year" "2004" "2010" "2015" ...
#> $ X.1 : chr "Series" "Threatened Species: Vertebrates (number)" "Threatened Species: Vertebrates (number)" "Threatened Species: Vertebrates (number)" ...
#> $ X.2 : chr "Value" "31" "31" "31" ...
#> $ X.3 : chr "Footnotes" "" "" "" ...
#> $ X.4 : chr "Source" "World Conservation Union (IUCN), Gland and Cambridge, IUCN Red List of Threatened Species publication, last acc"| __truncated__ "World Conservation Union (IUCN), Gland and Cambridge, IUCN Red List of Threatened Species publication, last acc"| __truncated__ "World Conservation Union (IUCN), Gland and Cambridge, IUCN Red List of Threatened Species publication, last acc"| __truncated__ ...1.2 Descarga de datos en formato .zip
Notar que en esta opción de download.file() no es necesario llamar el url con anterioridad. La función unzip se usa para descomprimir archivos. Para el ejemplo se bajará una base de datos Amazon de viajes de ciclistas en Nueva York
download.file(url='https://s3.amazonaws.com/tripdata/JC-202201-citibike-tripdata.csv.zip',destfile='file2.zip')
unzip("file2.zip")
file2 = read.csv("JC-202201-citibike-tripdata.csv", header=TRUE, sep = ",")
str(file2)
#> 'data.frame': 26762 obs. of 13 variables:
#> $ ride_id : chr "CA5837152804D4B5" "BA06A5E45B6601D2" "7B6827D7B9508D93" "6E5864EA6FCEC90D" ...
#> $ rideable_type : chr "electric_bike" "classic_bike" "classic_bike" "electric_bike" ...
#> $ started_at : chr "2022-01-26 18:50:39" "2022-01-28 13:14:07" "2022-01-10 19:55:13" "2022-01-26 07:54:57" ...
#> $ ended_at : chr "2022-01-26 18:51:53" "2022-01-28 13:20:23" "2022-01-10 20:00:37" "2022-01-26 07:55:22" ...
#> $ start_station_name: chr "12 St & Sinatra Dr N" "Essex Light Rail" "Essex Light Rail" "12 St & Sinatra Dr N" ...
#> $ start_station_id : chr "HB201" "JC038" "JC038" "HB201" ...
#> $ end_station_name : chr "12 St & Sinatra Dr N" "Essex Light Rail" "Essex Light Rail" "12 St & Sinatra Dr N" ...
#> $ end_station_id : chr "HB201" "JC038" "JC038" "HB201" ...
#> $ start_lat : num 40.8 40.7 40.7 40.8 40.8 ...
#> $ start_lng : num -74 -74 -74 -74 -74 ...
#> $ end_lat : num 40.8 40.7 40.7 40.8 40.8 ...
#> $ end_lng : num -74 -74 -74 -74 -74 ...
#> $ member_casual : chr "member" "member" "member" "member" ...1.3 Descarga de datos con programa RCurl
Esta es otra alternativa para la descarga de datos de intenet, usa la función getURL. Para archivos múltiples puede ser más rápida que download.file(). Se utilizará de ejemplo una base de datos de riesgo a la salud materna a partir de datos tomados usando dispositivos IoT (internet de las cosas) en Bangladesh
library(RCurl)
URL <- "http://archive.ics.uci.edu/ml/machine-learning-databases/00639/Maternal%20Health%20Risk%20Data%20Set.csv"
file3 <- getURL(URL)
file3 <- read.csv(textConnection(file3))
str(file3)
#> 'data.frame': 1014 obs. of 7 variables:
#> $ ï..Age : int 25 35 29 30 35 23 23 35 32 42 ...
#> $ SystolicBP : int 130 140 90 140 120 140 130 85 120 130 ...
#> $ DiastolicBP: int 80 90 70 85 60 80 70 60 90 80 ...
#> $ BS : num 15 13 8 7 6.1 7.01 7.01 11 6.9 18 ...
#> $ BodyTemp : num 98 98 100 98 98 98 98 102 98 98 ...
#> $ HeartRate : int 86 70 80 70 76 70 78 86 70 70 ...
#> $ RiskLevel : chr "high risk" "high risk" "high risk" "high risk" ...1.4 Descarga y lectura de archivos en formato xml
Los archivos xml (Extensible Markup Language) representan una de las formas más frecuentemente utilizadas para almacenar datos, que luego pueden ser leídos fácilmente por varios programas. Es un formato ampliamente utilizado en aplicaciones de internet. Se puede abrir, editar y crear un archivo XML con cualquier editor de texto. El formato XML es similar al HTML con la diferencia que XML sirve para transportar datos, mientras que HTML sirve para mostrarlos. A continuación, un ejemplo de la estructura de un catálogo de mascotas y de un catálogo de películas:


Se utilizarán la función GET() del programa httr para bajar los datos. Del programa XML se utilizará la función htmlParse() para “parsear” (recorrer y analizar la sintaxis de los datos) y la función xpathSApply() para buscar información específica dentro de los datos obtenidos. Se recomienda explorar esta última función con mayor profundidad dado que tiene múltiples utilidades. Como ejemplo se obtendrán datos de la página del perfil del autor del libro en Google Académico.
library(httr)
library(XML)
url = "https://scholar.google.es/citations?hl=es&user=0L1vFm8AAAAJ"
htm12 = GET(url)
content2 = content(htm12, as = "text")
parsedHtml = htmlParse(content2, asText=TRUE)
#parsedHtml
xpathSApply(parsedHtml, "//title", xmlValue)
#> [1] "Keilor Rojas-Jimenez - Google Académico"Desde R también se pueden realizar búsquedas directas en Google así como descargar automáticamente la información.
id = GET("http://google.com/", path = "search", query = list(q = "Costa Rica"))
content = content(id, as = "text")
parsed = htmlParse(content, asText=TRUE)
#parsed
xpathSApply(parsed, "//title", xmlValue)
#> [1] "Costa Rica - Buscar con Google"1.5 Web scraping
Es una técnica que utiliza programas para extraer información de sitios web. Los programas simulan la navegación de un humano en internet. El web scraping se aprovecha de la indexación de la información, sin embargo, se enfoca más en la transformación de datos sin estructura (p.e. HTML) en datos estructurados. Se usa p.e. para la comparación de precios en tiendas, monitoreo del clima y para obtener información relevante de páginas de internet.

Imagen ilustrativa tomada de (https://www.datalators.com/scraping-101/)
En el siguiente ejemplo se utilizará el programa rvest para extraer información contenida en tablas sobre el título de los artículos científicos más citados y el número de citas del autor de este libro en el perfil de Google Académico
library(rvest)
url <- 'https://scholar.google.es/citations?hl=es&user=0L1vFm8AAAAJ'
tables <- url %>%
read_html() %>%
html_nodes("table")
t01 <- tables %>%
purrr::pluck(1) %>%
html_table()
t01
#> # A tibble: 3 x 3
#> `` Total `Desde 2017`
#> <chr> <int> <int>
#> 1 Citas 1814 1546
#> 2 Índice h 18 16
#> 3 Índice i10 23 22
t02 <- tables %>%
purrr::pluck(2) %>%
html_table()
t02
#> # A tibble: 21 x 3
#> `` `` ``
#> <chr> <chr> <chr>
#> 1 TítuloOrdenarOrdenar por citasOrdenar por añ~ Cita~ Año
#> 2 Microplastic pollution increases gene exchan~ 309 2018
#> 3 Multilocus Sequence Analysis for Assessment ~ 199 2008
#> 4 Fungi in aquatic ecosystemsHP Grossart, S Va~ 186 2019
#> 5 Microplastics alter composition of fungal co~ 157 2017
#> 6 FungalTraits: a user-friendly traits databas~ 143 2021
#> 7 Integrating chytrid fungal parasites into pl~ 132 2017
#> 8 Aquatic fungi: targeting the forgotten in mi~ 92 2016
#> 9 Collateral effects of microplastic pollution~ 72 2019
#> 10 A ClC chloride channel homolog and ornithine~ 65 2005
#> # ... with 11 more rows1.6 Descarga y lectura de archivos en formato JSON
JSON (JavaScript Object Notation) es un formato de texto sencillo para representar e intercambiar datos estructurados en la sintaxis de objetos de JavaScript. Una de las ventajas de JSON sobre XML es que resulta mucho más sencillo de parsear. Es un formato más ligero para el almacenamiento de datos por lo que se usa en muchas API (Application Programming Interfaces). Los archivos JSON son almacenados en memoria en un formato de árbol.
A continuación, los diferentes tipos de valores en un archivo JSON:

Para la descarga de datos se utilizará la función fromJSON del paquete jsonlite. Se utilizará la función write.csv para guardar el archivo con formato .csv. Los datos de ejemplo proviene de una base de datos de registro de usuarios de diferentes programas de R
library(jsonlite)
data1 <- fromJSON("https://api.github.com/users/hadley/orgs")
names(data1)
#> [1] "login" "id"
#> [3] "node_id" "url"
#> [5] "repos_url" "events_url"
#> [7] "hooks_url" "issues_url"
#> [9] "members_url" "public_members_url"
#> [11] "avatar_url" "description"
data1$login
#> [1] "ggobi" "rstudio"
#> [3] "rstats" "ropensci"
#> [5] "rjournal" "r-dbi"
#> [7] "RConsortium" "tidyverse"
#> [9] "r-lib" "rstudio-education"
write.csv(data1, "data1json.csv")1.7 Descarga y lectura de archivos en formato MySQL
MySQL es un sistema de gestión de bases de datos relacional considerada como la base datos de código abierto más popular del mundo. MySQL es usado por muchos sitios web importantes como Wikipedia, Facebook, Twitter, Flickr y YouTube. Los datos están estructurados dentro de las bases de datos. Dentro de estas están las tablas y dentro de las tablas están los campos. Cada fila es considerada un registro.
En el siguiente ejemplo se utilizará el programa RMySQL para la descarga y lectura de datos del genoma del Manatee, entre muchos otros disponibles, que han sido generados por el Instituto de Genómica de la Universidad de California Santa Cruz. Las principales funciones para conectarse, explorar, bajar datos y desconectarse incluyen dbConnect, dbGetQuery(), dbListTables(), dbListFields(), dbReadTable() y dbDisconnect(). Recordar que al final se debe desconectar de la base de datos
library(RMySQL)
manatee = dbConnect(MySQL(), user = 'genome',db= 'triMan1',
host = 'genome-mysql.cse.ucsc.edu')
tables = dbListTables(manatee)
tables
#> [1] "all_mrna" "augustusGene"
#> [3] "bigFiles" "chainMm10"
#> [5] "chainMm10Link" "chromAlias"
#> [7] "chromInfo" "cpgIslandExt"
#> [9] "cpgIslandExtUnmasked" "cytoBandIdeo"
#> [11] "extNcbiRefSeq" "gap"
#> [13] "gbLoaded" "gc5BaseBw"
#> [15] "genscan" "gold"
#> [17] "grp" "hgFindSpec"
#> [19] "history" "microsat"
#> [21] "mrnaOrientInfo" "ncbiRefSeq"
#> [23] "ncbiRefSeqCds" "ncbiRefSeqCurated"
#> [25] "ncbiRefSeqLink" "ncbiRefSeqOther"
#> [27] "ncbiRefSeqPepTable" "ncbiRefSeqPredicted"
#> [29] "ncbiRefSeqPsl" "nestedRepeats"
#> [31] "netMm10" "rmsk"
#> [33] "seqNcbiRefSeq" "simpleRepeat"
#> [35] "tableDescriptions" "tableList"
#> [37] "trackDb" "ucscToINSDC"
#> [39] "ucscToRefSeq" "windowmaskerSdust"
#> [41] "xenoMrna" "xenoRefFlat"
#> [43] "xenoRefGene" "xenoRefSeqAli"
dbListFields(manatee,"all_mrna")
#> [1] "bin" "matches" "misMatches" "repMatches"
#> [5] "nCount" "qNumInsert" "qBaseInsert" "tNumInsert"
#> [9] "tBaseInsert" "strand" "qName" "qSize"
#> [13] "qStart" "qEnd" "tName" "tSize"
#> [17] "tStart" "tEnd" "blockCount" "blockSizes"
#> [21] "qStarts" "tStarts"
dbGetQuery(manatee, "select count(*) from all_mrna")# Para contabilizar el número de registros en la tabla
#> count(*)
#> 1 1297
dat = dbReadTable(manatee, "all_mrna")
head(dat)
#> bin matches misMatches repMatches nCount qNumInsert
#> 1 684 1042 5 0 0 0
#> 2 593 60 0 125 0 0
#> 3 593 37 0 0 0 0
#> 4 596 383 3 0 0 0
#> 5 604 50 0 51 0 0
#> 6 604 40 0 0 0 0
#> qBaseInsert tNumInsert tBaseInsert strand qName qSize
#> 1 0 4 4866 + AF055319 1047
#> 2 0 2 55156 - DQ323604 185
#> 3 0 0 0 - DQ323605 50
#> 4 0 1 282 + AY744134 391
#> 5 0 2 615 + DQ323633 101
#> 6 0 0 0 + DQ323634 50
#> qStart qEnd tName tSize tStart tEnd
#> 1 0 1047 JH594607 45942467 13030698 13036611
#> 2 0 185 JH594615 34412642 1094059 1149400
#> 3 0 37 JH594615 34412642 1149363 1149400
#> 4 5 391 JH594634 23036994 1531412 1532080
#> 5 0 101 JH594692 11983830 2534941 2535657
#> 6 10 50 JH594692 11983830 2535617 2535657
#> blockCount blockSizes qStarts
#> 1 5 361,169,166,240,111, 0,361,530,696,936,
#> 2 3 13,135,37, 0,13,148,
#> 3 1 37, 13,
#> 4 2 93,293, 5,98,
#> 5 3 10,51,40, 0,10,61,
#> 6 1 40, 10,
#> tStarts
#> 1 13030698,13033153,13034913,13035233,13036500,
#> 2 1094059,1095700,1149363,
#> 3 1149363,
#> 4 1531412,1531787,
#> 5 2534941,2535319,2535617,
#> 6 2535617,
dbDisconnect(manatee)
#> [1] TRUE1.8 Descarga y lectura de datos del NCBI
El programa rentrez que proporciona una interfaz a la API del Centro Nacional de Información de Biotecnología de los Estados Unidos (NCBI), lo que permite a los usuarios buscar bases de datos de secuencias como GenBank y de publicaciones PubMed. También permite extraer y procesar los resultados de las búsquedas. En el ejemplo se buscarán una serie de secuencias de nucleótidos y su información asociada de acuerdo a su número de acceso usando la función entrez_fetch()
library("rentrez")
entrez_dbs()#lista de bases de datos
#> [1] "pubmed" "protein" "nuccore"
#> [4] "ipg" "nucleotide" "structure"
#> [7] "genome" "annotinfo" "assembly"
#> [10] "bioproject" "biosample" "blastdbinfo"
#> [13] "books" "cdd" "clinvar"
#> [16] "gap" "gapplus" "grasp"
#> [19] "dbvar" "gene" "gds"
#> [22] "geoprofiles" "homologene" "medgen"
#> [25] "mesh" "nlmcatalog" "omim"
#> [28] "orgtrack" "pmc" "popset"
#> [31] "proteinclusters" "pcassay" "protfam"
#> [34] "pccompound" "pcsubstance" "seqannot"
#> [37] "snp" "sra" "taxonomy"
#> [40] "biocollections" "gtr"
entrez_db_searchable("nucleotide")#argumentos de búsqueda
#> Searchable fields for database 'nuccore'
#> ALL All terms from all searchable fields
#> UID Unique number assigned to each sequence
#> FILT Limits the records
#> WORD Free text associated with record
#> TITL Words in definition line
#> KYWD Nonstandardized terms provided by submitter
#> AUTH Author(s) of publication
#> JOUR Journal abbreviation of publication
#> VOL Volume number of publication
#> ISS Issue number of publication
#> PAGE Page number(s) of publication
#> ORGN Scientific and common names of organism, and all higher levels of taxonomy
#> ACCN Accession number of sequence
#> PACC Does not include retired secondary accessions
#> GENE Name of gene associated with sequence
#> PROT Name of protein associated with sequence
#> ECNO EC number for enzyme or CAS registry number
#> PDAT Date sequence added to GenBank
#> MDAT Date of last update
#> SUBS CAS chemical name or MEDLINE Substance Name
#> PROP Classification by source qualifiers and molecule type
#> SQID String identifier for sequence
#> GPRJ BioProject
#> SLEN Length of sequence
#> FKEY Feature annotated on sequence
#> PORG Scientific and common names of primary organism, and all higher levels of taxonomy
#> COMP Component accessions for an assembly
#> ASSM Assembly
#> DIV Division
#> STRN Strain
#> ISOL Isolate
#> CULT Cultivar
#> BRD Breed
#> BIOS BioSample
acc <- c("U42478.1", "GU810145.1", "EF200529.1", "KX354828.1", "JQ796369.1", "HQ888719.1", "HQ901774.1", "GU810144.1", "KX354828.1", "NG_017165.1", "JF414188.1", "GU810144.1", "JX869391.1", "KJ563218.1", "KJ563218.1", "GU810145.1", "KJ952219.1", "AY665775.1", "KT595127.1", "KX354828.1", "KF036709.1", "NG_017165.1", "JX869391.1")
seqs <- entrez_fetch(db="nuccore", id=acc, rettype="fasta")
write(seqs, file="my_seqs.fasta") # Revisar archivo con secuencias en directorioSe puede buscar la información asociada a cada número de acceso.
info <- entrez_fetch(db="nuccore", id=acc, rettype="gb")
write(info, file="info_seqs.txt") # Revisar archivo en carpeta de trabajoTambién se utilizará la función entrez_search() para buscar la información taxonómica de una especie.
Tt <- entrez_search(db="taxonomy", term="(Tetrahymena thermophila[ORGN]) AND Species[RANK]")
tax_rec <- entrez_fetch(db="taxonomy", id=Tt$ids, rettype="xml", parsed=TRUE)
tax_list <- XML::xmlToList(tax_rec)
tax_list$Taxon$GeneticCode
#> $GCId
#> [1] "6"
#>
#> $GCName
#> [1] "Ciliate Nuclear; Dasycladacean Nuclear; Hexamita Nuclear"
tt_lineage <- tax_rec["//LineageEx/Taxon/ScientificName"]
XML::xpathSApply(tax_rec, "//LineageEx/Taxon/ScientificName", XML::xmlValue)
#> [1] "cellular organisms" "Eukaryota"
#> [3] "Sar" "Alveolata"
#> [5] "Ciliophora" "Intramacronucleata"
#> [7] "Oligohymenophorea" "Hymenostomatida"
#> [9] "Tetrahymenina" "Tetrahymenidae"
#> [11] "Tetrahymena"1.9 Estandarización de formato de fechas con programa Lubridate
Uno de los problemas más comunes al trabajar con bases de datos de diferentes fuentes constituye el formato de las fechas. Lubridate es un paquete R que facilita el trabajo con fechas y horas. Las funciones de análisis manejan una amplia variedad de formatos y separadores, lo que simplifica el proceso de análisis. Incluso se pueden realizar operaciones matemáticas con fechas.
library("lubridate")
date()
#> [1] "Mon Sep 26 20:16:36 2022"
now()
#> [1] "2022-09-26 20:16:36 CST"
ymd("20190317")
#> [1] "2019-03-17"
mdy("03-17-2019")
#> [1] "2019-03-17"
dmy("17/03/2019")
#> [1] "2019-03-17"
ymd("20190317") + years(1:3)
#> [1] "2020-03-17" "2021-03-17" "2022-03-17"
ymd("20190317") + months(0:6)
#> [1] "2019-03-17" "2019-04-17" "2019-05-17" "2019-06-17"
#> [5] "2019-07-17" "2019-08-17" "2019-09-17"1.10 Expresiones regulares
Una expresión regular (regex), es una secuencia que forma un patrón de búsqueda en cadenas de caracteres u operaciones de sustituciones. Son fundamentales en la limpieza y edición de texto. Es una herramienta de primera mano de toda persona que trabaja en ciencia de datos.

En la función sub() se sustituye el primer elemento que está entre “comillas” por el segundo elemento entre “comillas”. En la función gsub() la sustitución actual de manera más global, siguiendo el siguiente orden: gsub(patrón, remplazo,nombre_archivo).
test = "Esta_es_una_prueba"
sub("_","", test)# sustituye el primer elemento que está entre "comillas" por el segundo elemento entre "comillas".
#> [1] "Estaes_una_prueba"
gsub("_","", test)# similar a sub pero actua a nivel global
#> [1] "Estaesunaprueba"La función grep() permite detectar y llamar diferentes patrones en una tabla o un texto.
data <- data.frame(values = c(91, 92, 108, 104, 87, 91, 91, 97, 81, 98),
names = c("fee-", "fi", "fo-", "fum-", "foo-", "foo1234-", "123foo-", "fum-", "fum-", "fum-"))
grep("foo",data$names, value=T)
#> [1] "foo-" "foo1234-" "123foo-"
data$values[grep("foo",data$names)]
#> [1] 87 91 91Se pueden hacer pequeñas funciones donde se procesa información solicitada sobre el patrón y se arroja un resultado.
txt <- c("arm","foot","lefroo", "bafoobar")
if(length(i <- grep("foo", txt)))
cat("'foo' aparece al menos una vez en\n\t", txt, "\n")
#> 'foo' aparece al menos una vez en
#> arm foot lefroo bafoobarTambién se puede hacer un análisis de vectores lógicos sobre una lista o un texto.
1.11 Resumen de datos
Antes de empezar a realizar cualquier análisis de datos formal es importante tener un acercamiento rápido para conocer la estructura de la base de datos, la cantidad de filas y columnas, los tipos de variables, etc. Para esto se puede usar la función str o la función glimpsedel paquete tidyr. La base de datos del ejemplo corresponde mediciones de la densidad mineral del hueso espinal en adolescentes de Estados Unidos tomada de un repositorio de bases de datos para aprendizaje de estadística de la Universidad de Standford.
library(Hmisc)
library(ggplot2)
library(tibble)
download.file(url='https://web.stanford.edu/~hastie/ElemStatLearn/datasets/spnbmd.csv',
destfile='bone.csv')
bone = read.csv2("bone.csv", header = TRUE, sep = ",", dec = ".")
head(bone)
#> idnum ethnic age sex spnbmd
#> 1 1 White 11.2 mal 0.719
#> 2 1 White 12.2 mal 0.732
#> 3 1 White 13.2 mal 0.776
#> 4 1 White 14.3 mal 0.781
#> 5 2 White 12.7 mal 0.620
#> 6 2 White 13.8 mal 0.627
str(bone)
#> 'data.frame': 1003 obs. of 5 variables:
#> $ idnum : int 1 1 1 1 2 2 2 2 3 3 ...
#> $ ethnic: chr "White" "White" "White" "White" ...
#> $ age : num 11.2 12.2 13.2 14.3 12.7 13.8 14.8 15.8 10.9 11.9 ...
#> $ sex : chr "mal" "mal" "mal" "mal" ...
#> $ spnbmd: num 0.719 0.732 0.776 0.781 0.62 0.627 0.759 0.79 0.641 0.622 ...
glimpse(bone)
#> Rows: 1,003
#> Columns: 5
#> $ idnum <int> 1, 1, 1, 1, 2, 2, 2, 2, 3, 3, 3, 3, 4, 4, 4~
#> $ ethnic <chr> "White", "White", "White", "White", "White"~
#> $ age <dbl> 11.2, 12.2, 13.2, 14.3, 12.7, 13.8, 14.8, 1~
#> $ sex <chr> "mal", "mal", "mal", "mal", "mal", "mal", "~
#> $ spnbmd <dbl> 0.719, 0.732, 0.776, 0.781, 0.620, 0.627, 0~La función summary() es útil para generar un primer resumen estadístico. Se pueden generar histogramas y gráficos de cajas con el paquete ggplot. Algunas veces es útil también subdividir las variables en rangos (cuantiles), calculados con la función cut2() del programa Hmisc. También se pueden realizar algunos test estadísticos.
summary(bone)
#> idnum ethnic age
#> Min. : 1.0 Length:1003 Min. : 8.80
#> 1st Qu.: 84.5 Class :character 1st Qu.:12.80
#> Median :181.0 Mode :character Median :15.70
#> Mean :189.9 Mean :16.32
#> 3rd Qu.:287.5 3rd Qu.:19.45
#> Max. :429.0 Max. :26.20
#> sex spnbmd
#> Length:1003 Min. :0.5360
#> Class :character 1st Qu.:0.7995
#> Mode :character Median :0.9650
#> Mean :0.9476
#> 3rd Qu.:1.0705
#> Max. :1.4430
ggplot(bone, aes(x=spnbmd)) + geom_histogram(binwidth=0.05)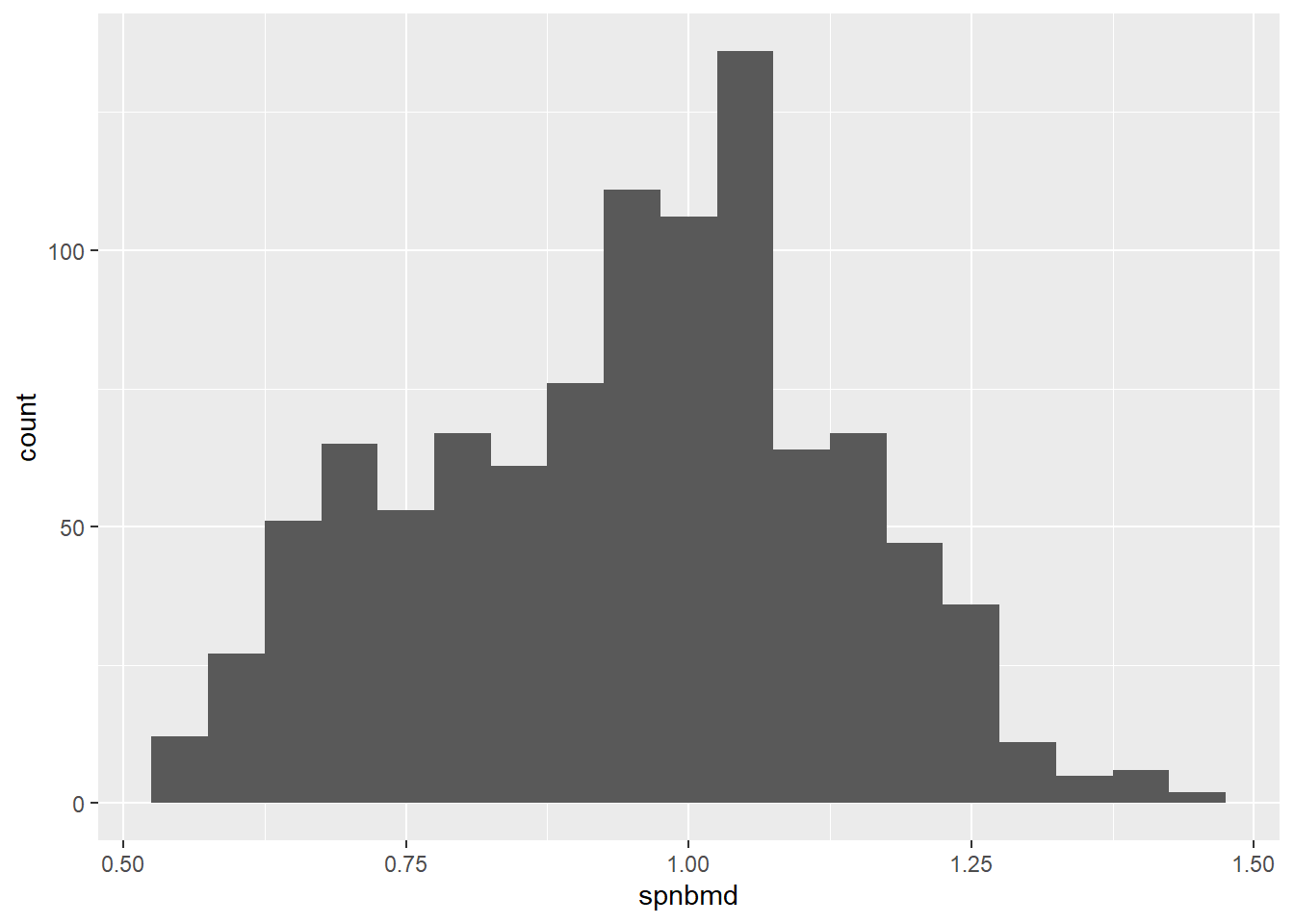
Figure 1.1: 1
ggplot(bone, aes(factor(sex), spnbmd)) + geom_boxplot()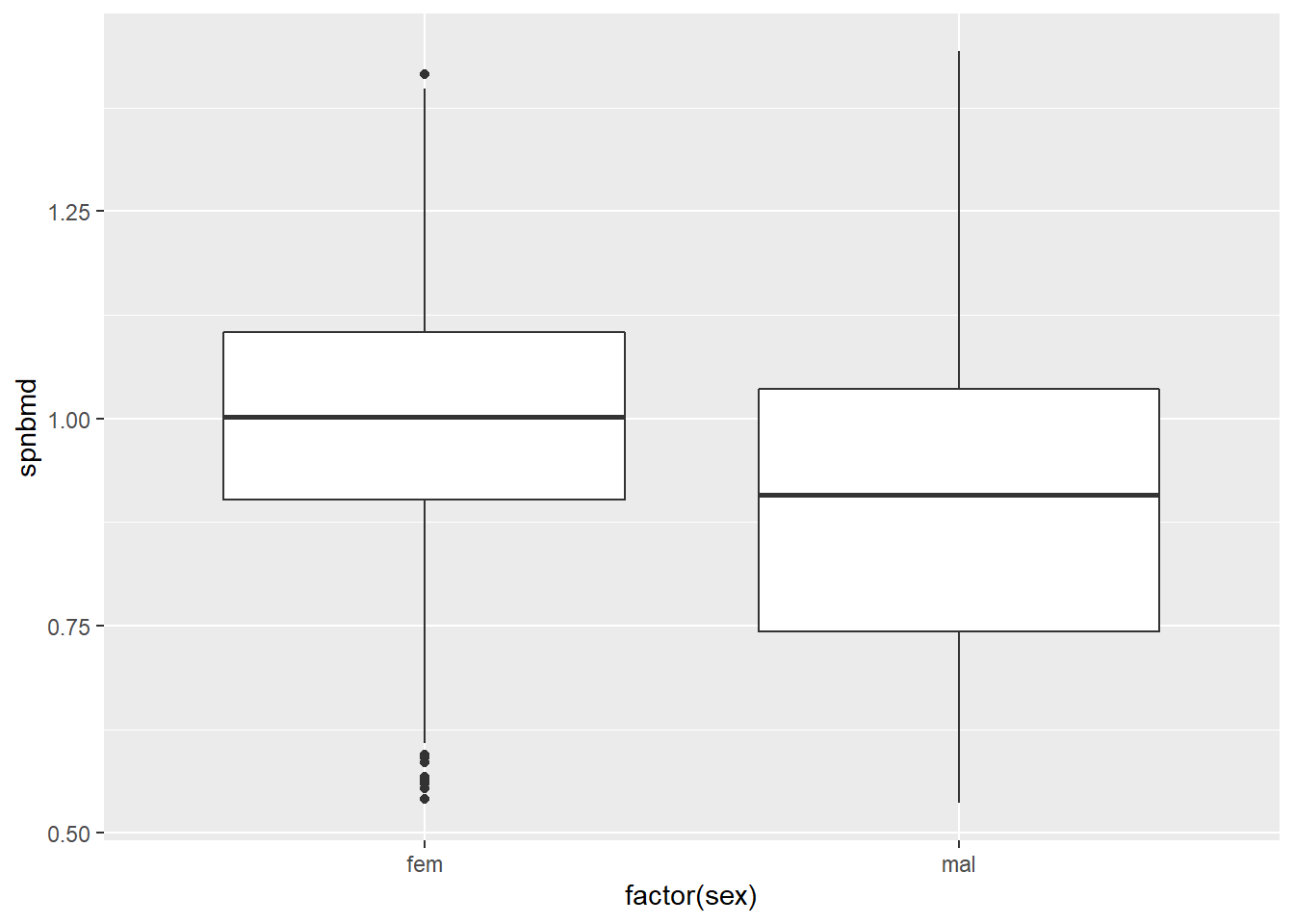
Figure 1.2: 1
age3 <- cut2(bone$age,g=3)
table(age3)
#> age3
#> [ 8.8,13.8) [13.8,18.3) [18.3,26.2]
#> 340 339 324
ggplot(bone, aes(factor(age3), spnbmd)) + geom_boxplot()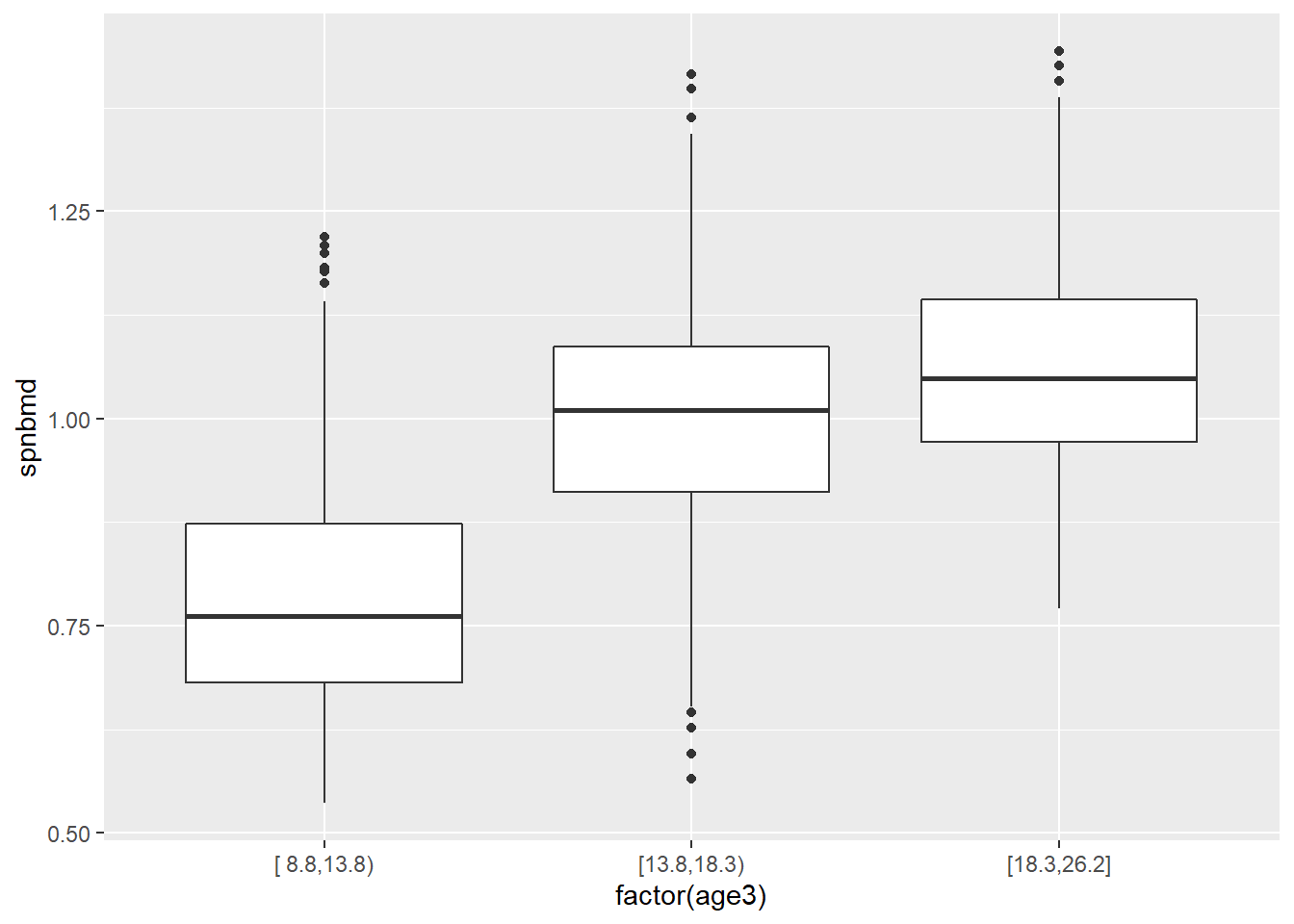
Figure 1.3: 1
ggplot(bone, aes(factor(ethnic), spnbmd)) + geom_boxplot()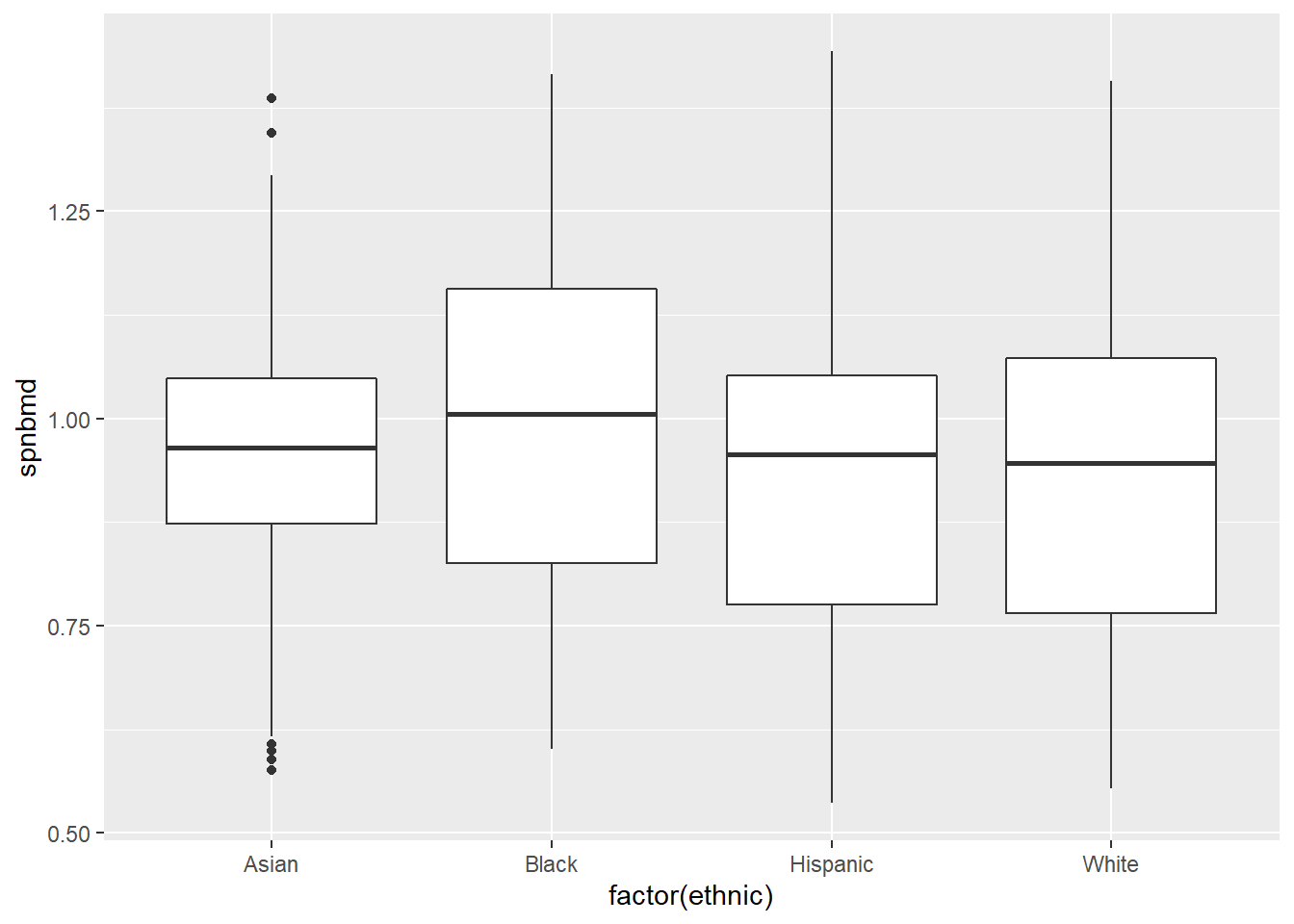
Figure 1.4: 1
kruskal.test(spnbmd ~ ethnic, data = bone)
#>
#> Kruskal-Wallis rank sum test
#>
#> data: spnbmd by ethnic
#> Kruskal-Wallis chi-squared = 19.093, df = 3, p-value
#> = 0.0002616
pairwise.wilcox.test(bone$spnbmd, bone$ethnic, p.adjust.method="bonferroni")
#>
#> Pairwise comparisons using Wilcoxon rank sum test with continuity correction
#>
#> data: bone$spnbmd and bone$ethnic
#>
#> Asian Black Hispanic
#> Black 0.07944 - -
#> Hispanic 0.93399 0.00133 -
#> White 1.00000 0.00067 1.00000
#>
#> P value adjustment method: bonferroni1.12 Descarga de datos con el programa readr
En el manejo cotidiano de datos en R se utilizan muchas herramientas, sin embargo, una de las más importantes son los programas de Tidyverse. Lo que se denomina el universo Tidyverse es en realidad una colección de paquetes R diseñados para la ciencia de datos que comparten una filosofía de diseño, gramática y estructuras de datos subyacentes.
Dentro de este conjunto de paquetes, readr se especializa en la obtención de datos de archivos separados por comas (csv), separados por tabulaciones (tsv) o de ancho fijo (fwf) en R. Está diseñado para analizar de manera flexible y rápida muchos tipos de datos. Es un paquete con muchas funcionalidades y, por tanto, una de las herramientas más utilizadas. Notar que la función read_csv() se utiliza para archivos .csv separados por coma (,) la función read_csv2() se utiliza para archivos separados por punto y coma (;), mientras que la función read_delim permite leer otros delimitadores particulares como por ejemplo el “|”.
Es importante mencionar que readr transforma los data.frame en tibble. Para quien ha estado usando R , probablemente esté familiarizado con el data.frame. Se podría decir que los tibbles son una versión actualizada y elegante del data.frame que funcionan muy bien en tidyverse.
library(readr)
url <- "http://data.un.org/_Docs/SYB/CSV/SYB64_313_202110_Threatened%20Species.csv"
read_csv(file = url)# Notar que primera fila causa distorsión de los datos
#> # A tibble: 6,921 x 7
#> T23 `Threatened sp~` ...3 ...4 ...5 ...6 ...7
#> <chr> <chr> <chr> <chr> <chr> <chr> <chr>
#> 1 Region/Co~ <NA> Year Seri~ Value Foot~ Sour~
#> 2 4 Afghanistan 2004 Thre~ 31 <NA> Worl~
#> 3 4 Afghanistan 2010 Thre~ 31 <NA> Worl~
#> 4 4 Afghanistan 2015 Thre~ 31 <NA> Worl~
#> 5 4 Afghanistan 2018 Thre~ 34 <NA> Worl~
#> 6 4 Afghanistan 2019 Thre~ 33 <NA> Worl~
#> 7 4 Afghanistan 2020 Thre~ 33 <NA> Worl~
#> 8 4 Afghanistan 2021 Thre~ 38 <NA> Worl~
#> 9 4 Afghanistan 2004 Thre~ 1 <NA> Worl~
#> 10 4 Afghanistan 2010 Thre~ 1 <NA> Worl~
#> # ... with 6,911 more rowsEl parámetro skip se usa para eliminar filas que a veces es información suplementaria que contienen las bases de datos pero que distorcionan su lectura. Notar también que un aspecto positivo de la función read_csves que permite identificar la clase de dato para cada columna. El parámetro n_max permite especificar el número de filas que se quieren seleccionar. El parámetro na permite definir la información dentro de las celdas que será tratada como valores perdidos.
library(readr)
read_csv(file=url,skip=1, n_max = 10, na= "World Conservation Union (IUCN), Gland and Cambridge, IUCN Red List of Threatened Species publication, last accessed June 2021.")
#> # A tibble: 10 x 7
#> `Region/Country/Area` ...2 Year Series Value Footnotes
#> <dbl> <chr> <dbl> <chr> <dbl> <lgl>
#> 1 4 Afgha~ 2004 Threa~ 31 NA
#> 2 4 Afgha~ 2010 Threa~ 31 NA
#> 3 4 Afgha~ 2015 Threa~ 31 NA
#> 4 4 Afgha~ 2018 Threa~ 34 NA
#> 5 4 Afgha~ 2019 Threa~ 33 NA
#> 6 4 Afgha~ 2020 Threa~ 33 NA
#> 7 4 Afgha~ 2021 Threa~ 38 NA
#> 8 4 Afgha~ 2004 Threa~ 1 NA
#> 9 4 Afgha~ 2010 Threa~ 1 NA
#> 10 4 Afgha~ 2015 Threa~ 2 NA
#> # ... with 1 more variable: Source <lgl>La función View() se utiliza para desplegar la tabla generada detro de una nueva ventana de R Studio, la función .Last.value llama el resultado del último comando ejecutado.
View(.Last.value)Otra ventaja del programa readr es que se puede hacer edición sobre el archivo que se está bajando o leyendo. Por ejemplo, se puede seleccionar y cambiar de nombre a las columnas con el parámetro col_names así como cambiar la clase de las columnas con el parámetro col_types.
library(readr)
amenazado= read_csv(file=url,
skip=2,
n_max=10,
col_names = c("Region", "Pais", "Año", "Amenaza","Valor","Notas", "Fuente"),
col_types = cols(Año =col_factor()))1.13 Descarga de datos con el programa readxl
El paquete readxl permite la transferencia de datos de Excel a R. Este programa también forma parte del ambiente Tidyverse. En comparación con muchos de los paquetes existentes readxl no tiene dependencias externas, por lo que es fácil de instalar y usar en todos los sistemas operativos. Está diseñado para trabajar con datos tabulares. La función read_excel() admite tanto los formatos .xls y .xlsx. Se utilizarán los archivos de ejemplo del programa
library(readxl)
readxl_example()# para ver los archivos disponibles
#> [1] "clippy.xls" "clippy.xlsx" "datasets.xls"
#> [4] "datasets.xlsx" "deaths.xls" "deaths.xlsx"
#> [7] "geometry.xls" "geometry.xlsx" "type-me.xls"
#> [10] "type-me.xlsx"
data <- readxl_example("datasets.xlsx")
excel_sheets(data) # para ver los nombres de las hojas de cálculo dentro del archivo
#> [1] "iris" "mtcars" "chickwts" "quakes"
read_excel(data)# llama por defecto los datos de la primera hoja.
#> # A tibble: 150 x 5
#> Sepal.Length Sepal.Width Petal.Length Petal.Width Species
#> <dbl> <dbl> <dbl> <dbl> <chr>
#> 1 5.1 3.5 1.4 0.2 setosa
#> 2 4.9 3 1.4 0.2 setosa
#> 3 4.7 3.2 1.3 0.2 setosa
#> 4 4.6 3.1 1.5 0.2 setosa
#> 5 5 3.6 1.4 0.2 setosa
#> 6 5.4 3.9 1.7 0.4 setosa
#> 7 4.6 3.4 1.4 0.3 setosa
#> 8 5 3.4 1.5 0.2 setosa
#> 9 4.4 2.9 1.4 0.2 setosa
#> 10 4.9 3.1 1.5 0.1 setosa
#> # ... with 140 more rowsEl parámetro sheet permite escoger la hoja del archivo excel de la cual se extraerán los datos y el parámetro cell_cols permite seleccionar las columnas.
data2 <- read_excel(data,sheet= "quakes",range = cell_cols(1:4))
data2
#> # A tibble: 1,000 x 4
#> lat long depth mag
#> <dbl> <dbl> <dbl> <dbl>
#> 1 -20.4 182. 562 4.8
#> 2 -20.6 181. 650 4.2
#> 3 -26 184. 42 5.4
#> 4 -18.0 182. 626 4.1
#> 5 -20.4 182. 649 4
#> 6 -19.7 184. 195 4
#> 7 -11.7 166. 82 4.8
#> 8 -28.1 182. 194 4.4
#> 9 -28.7 182. 211 4.7
#> 10 -17.5 180. 622 4.3
#> # ... with 990 more rowsTambién se pueden seleccionar un número de filas definido
data3 <- read_excel(data, range = cell_rows(1:10))
data3
#> # A tibble: 9 x 5
#> Sepal.Length Sepal.Width Petal.Length Petal.Width Species
#> <dbl> <dbl> <dbl> <dbl> <chr>
#> 1 5.1 3.5 1.4 0.2 setosa
#> 2 4.9 3 1.4 0.2 setosa
#> 3 4.7 3.2 1.3 0.2 setosa
#> 4 4.6 3.1 1.5 0.2 setosa
#> 5 5 3.6 1.4 0.2 setosa
#> 6 5.4 3.9 1.7 0.4 setosa
#> 7 4.6 3.4 1.4 0.3 setosa
#> 8 5 3.4 1.5 0.2 setosa
#> 9 4.4 2.9 1.4 0.2 setosaTambién se puede pueden definir un rango de celdas específico, tal cual se estuvieran seleccionando con mouse. Incluso se puede editar el nombre de las columnas para que sea en mayúsculas con el parámetro .name_repair.
data4 <- read_excel(data,sheet= "quakes",range = "A1:D10",.name_repair = toupper)
data4
#> # A tibble: 9 x 4
#> LAT LONG DEPTH MAG
#> <dbl> <dbl> <dbl> <dbl>
#> 1 -20.4 182. 562 4.8
#> 2 -20.6 181. 650 4.2
#> 3 -26 184. 42 5.4
#> 4 -18.0 182. 626 4.1
#> 5 -20.4 182. 649 4
#> 6 -19.7 184. 195 4
#> 7 -11.7 166. 82 4.8
#> 8 -28.1 182. 194 4.4
#> 9 -28.7 182. 211 4.7La función read_excel() adivinará los tipos de datos en las columnas de forma predeterminada, o también se pueden proporcionar explícitamente a través del argumento col_types. Dado que provienen de Excel, cada celda puede ser: vacia < booleana < numerica < texto, donde las fechas son un tipo de celda numérica
data5 <- read_excel(readxl_example("deaths.xlsx"), range = cell_rows(5:15))
data5
#> # A tibble: 10 x 6
#> Name Profession Age `Has kids` `Date of birth`
#> <chr> <chr> <dbl> <lgl> <dttm>
#> 1 David Bo~ musician 69 TRUE 1947-01-08 00:00:00
#> 2 Carrie F~ actor 60 TRUE 1956-10-21 00:00:00
#> 3 Chuck Be~ musician 90 TRUE 1926-10-18 00:00:00
#> 4 Bill Pax~ actor 61 TRUE 1955-05-17 00:00:00
#> 5 Prince musician 57 TRUE 1958-06-07 00:00:00
#> 6 Alan Ric~ actor 69 FALSE 1946-02-21 00:00:00
#> 7 Florence~ actor 82 TRUE 1934-02-14 00:00:00
#> 8 Harper L~ author 89 FALSE 1926-04-28 00:00:00
#> 9 Zsa Zsa ~ actor 99 TRUE 1917-02-06 00:00:00
#> 10 George M~ musician 53 FALSE 1963-06-25 00:00:00
#> # ... with 1 more variable: `Date of death` <dttm>También se puede cambiar el nombre a las columnas con el argumento col_names, usar el parámetro skip para brincarse un número de filas y el parámetro n_max para seleccionar un número máximo de filas que se mostrarán.
data6 <- read_excel(data,sheet= "quakes", skip=1,
col_names = c("latitud","longitud","profundidad", "magnitud", "estaciones"),
col_types =c("numeric", "guess", "guess", "numeric","guess"),
n_max=3)
data6
#> # A tibble: 3 x 5
#> latitud longitud profundidad magnitud estaciones
#> <dbl> <dbl> <dbl> <dbl> <dbl>
#> 1 -20.4 182. 562 4.8 41
#> 2 -20.6 181. 650 4.2 15
#> 3 -26 184. 42 5.4 431.14 Formatos propios de R
Muchas veces, luego de haber hecho la lectura y edición de la tablas de datos, es conveniente mantener información asociada, como por ejemplo las definiociones de las clases de las columnas como factores o número enteros. Esto se hace mediante los formatos propios de R.
Existen dos tipos de formatos:
- El formato Rds se utiliza para guardar un solo objeto
- El formato RData (Rda) se utiliza para guardar varios objetos juntos.
De esta manera, continuando con los datos del apartado anterior, dos o más archivos se podrían salvar así:
save(data2, data3,file = "datosjuntos.RData")Para leerlos se utiliza la función load.
load("datosjuntos.RData")
head(data2)
#> # A tibble: 6 x 4
#> lat long depth mag
#> <dbl> <dbl> <dbl> <dbl>
#> 1 -20.4 182. 562 4.8
#> 2 -20.6 181. 650 4.2
#> 3 -26 184. 42 5.4
#> 4 -18.0 182. 626 4.1
#> 5 -20.4 182. 649 4
#> 6 -19.7 184. 195 4Para grabar un archivo como .RDS con la función write_rds(). Esta última función es más útil cuando se trabaja en flujos continuos de datos.También permite guardar los metadados asociados.
write_rds(data3, "data3.rds")Para importar los datos se utiliza la función readRDS()
readRDS("data3.rds")
#> # A tibble: 9 x 5
#> Sepal.Length Sepal.Width Petal.Length Petal.Width Species
#> <dbl> <dbl> <dbl> <dbl> <chr>
#> 1 5.1 3.5 1.4 0.2 setosa
#> 2 4.9 3 1.4 0.2 setosa
#> 3 4.7 3.2 1.3 0.2 setosa
#> 4 4.6 3.1 1.5 0.2 setosa
#> 5 5 3.6 1.4 0.2 setosa
#> 6 5.4 3.9 1.7 0.4 setosa
#> 7 4.6 3.4 1.4 0.3 setosa
#> 8 5 3.4 1.5 0.2 setosa
#> 9 4.4 2.9 1.4 0.2 setosa
head(data3)
#> # A tibble: 6 x 5
#> Sepal.Length Sepal.Width Petal.Length Petal.Width Species
#> <dbl> <dbl> <dbl> <dbl> <chr>
#> 1 5.1 3.5 1.4 0.2 setosa
#> 2 4.9 3 1.4 0.2 setosa
#> 3 4.7 3.2 1.3 0.2 setosa
#> 4 4.6 3.1 1.5 0.2 setosa
#> 5 5 3.6 1.4 0.2 setosa
#> 6 5.4 3.9 1.7 0.4 setosa1.15 Lectura de otros tipos de datos
En este capítulo se presentó algunos programas y comandos para la lectura de diferentes tipos de datos, sin embargo, es natural que existan muchos otros tipos de datos y programas para bajarlos p.e. los de sistemas de información geográfica, imágenes, audio y video. Por esto se presenta a continuación una lista de recursos adicionales que podrían ser útiles para esos formatos.
Sistemas de información geográfica
- raster (https://cran.r-project.org/web/packages/raster/index.html)
- rdgal (https://cran.r-project.org/web/packages/rgdal/index.html )
- rgeos (https://cran.r-project.org/web/packages/rgeos/index.html )
- marmap (https://cran.r-project.org/web/packages/marmap/index.html)
Imágenes
- magick(https://cran.r-project.org/web/packages/magick/index.html)
- imager (https://cran.r-project.org/web/packages/imager/index.html )
- jpeg (https://cran.r-project.org/web/packages/jpeg/index.html)
- png (https://cran.r-project.org/web/packages/png/index.html)
Audio
- tuneR (https://cran.r-project.org/web/packages/tuneR/index.html)
- seewave (https://cran.r-project.org/web/packages/seewave/index.html)
Video
1.16 Bases de datos de bases de datos
Las bases de datos de bases de datos constituyen un recurso muy valioso porque hacen de acceso público los datos generados por las propias instituciones o compilados de diversas fuentes. A continuación una lista de links:
- (http://data.un.org/)
- (https://www.eea.europa.eu/data-and-maps)
- (http://storage.googleapis.com/books/ngrams/books/datasetsv2.html)
- (https://www.cancerimagingarchive.net/)
- (https://labrosa.ee.columbia.edu/millionsong/)
- (https://www.reddit.com/r/datasets/)
- (https://arxiv.org/help/bulk_data_s3)
- (http://netsg.cs.sfu.ca/youtubedata/)
- (http://www.face-rec.org/databases/)
- (https://web.stanford.edu/~hastie/ElemStatLearn//data.html )
- (http://crcns.org/data-sets)
- (https://www.opensciencedatacloud.org/)
- (https://aws.amazon.com/es/opendata/open-data-sponsorship-program/)
- (https://www.google.com/publicdata/directory)
- (http://archive.ics.uci.edu/ml/datasets.php)
1.17 Recursos complementarios
- https://jhudatascience.org/tidyversecourse/get-data.html
- https://benwhalley.github.io/just-enough-r/index.html
- https://static-bcrf.biochem.wisc.edu/courses/Tabular-data-analysis-with-R-and-Tidyverse/book/
- https://r4ds.had.co.nz/index.html
- https://www.institutomora.edu.mx/testU/SitePages/martinpaladino/manipulacion_de_datos_con_r_dplyr_y_tidyr.html
- https://www.uv.es/pjperez/curso_R/index.html
- https://www.rstudio.com/resources/cheatsheets/