Part 3 My Code
I will admit: I have used packages and techniques that go beyond the scope of BS0004.
For this reason, there are many sections to this .html document that you’re currently reading. In those sections, I attempt to elaborate on parts of the workflow that are not covered by professors Wilson or Jarkko.
3.1 Setting Up the Working Environment
I first set the working directory of my current R session to my laptop’s “Downloads” folder:
setwd("C:/Users/Kevin/Downloads")I then loaded in the following packages:
library(tidyverse) ; library(pheatmap) ; library(ggplot2)
library(ROCR) ; library(caret) ; library(e1071)
library(gprofiler2) ; library(corrplot) ; library(vip)I will not elaborate on what the ggplot2 or what the pheatmap package does. These have already been explained in lectures 5 and onwards.
Nonetheless:
- The
tidyversepackages are a collection packages authored by Hadley Wickham that obey his tidy data principle. While not technically a part of thetidyverse, callinglibrary(tidyverse)loads in themagrittrpackage. The biggest boon ofmagrittris the pipe%>%. Thetidyversepackages are (in no particular order):stringr: this package is for string processing.tidyr: this is used for tidying up data.dplyr: this is used for data manipulation.readr: this is used for reading in data.purrr: this is used for functional programming.
- I used the
ROCRpackage to plot the ROC curves of the various supervised clustering models and calculate their AUCs. - I used the
caretpackage to not only train the majority of the supervised clustering models, but also perform k-fold cross validation. - I used the
e1071package to create a linear support vector machine. - I used the
gprofiler2package to perform GO term analysis. This package has been endorsed by the Gene Ontology. - I used the
corrplotpackage to generate aesthetically-pleasing correlation matrices. - I used the
vippackage to generate variable importance plots.
3.2 Exploring the Data
While I am not sure what has been done to the data to clean it (do ask Sean - he was the one who sent me the data in a .csv file), I began by reading the data into my current R session:
data <- read.csv("original idea.csv")I then decided to transpose the original data frame and save the transposed data frame in a variable called transposed. While I was at this, I also changed the row and column names of transposed to the column and row names of data respectively:
row <- data[, 1] ; data <- data[, -1] ; col <- colnames(data)
transposed <- data %>% t() %>% as.data.frame()
colnames(transposed) <- row ; rownames(transposed) <- colI also created a new variable in transposed called Status: a factor that states whether or not a sample (i.e., patient) is a control or has been treated:
transposed$Status <- c(rep("Control", 2), rep("Treated", 4), rep("Control", 10), rep("Treated", 8))
transposed$Status <- as.factor(transposed$Status)In the next several sections, I then performed some exploratory data analysis prior to actually building the supervised classifiers. I’ve also constructed numerous plots during this step of the workflow.
3.2.1 A heatmap using pheatmap()
I made a heatmap using all the genes as suggested by Sean:
pheatmap(transposed[, 1:(ncol(transposed) - 1)], scale = "column", show_colnames = FALSE)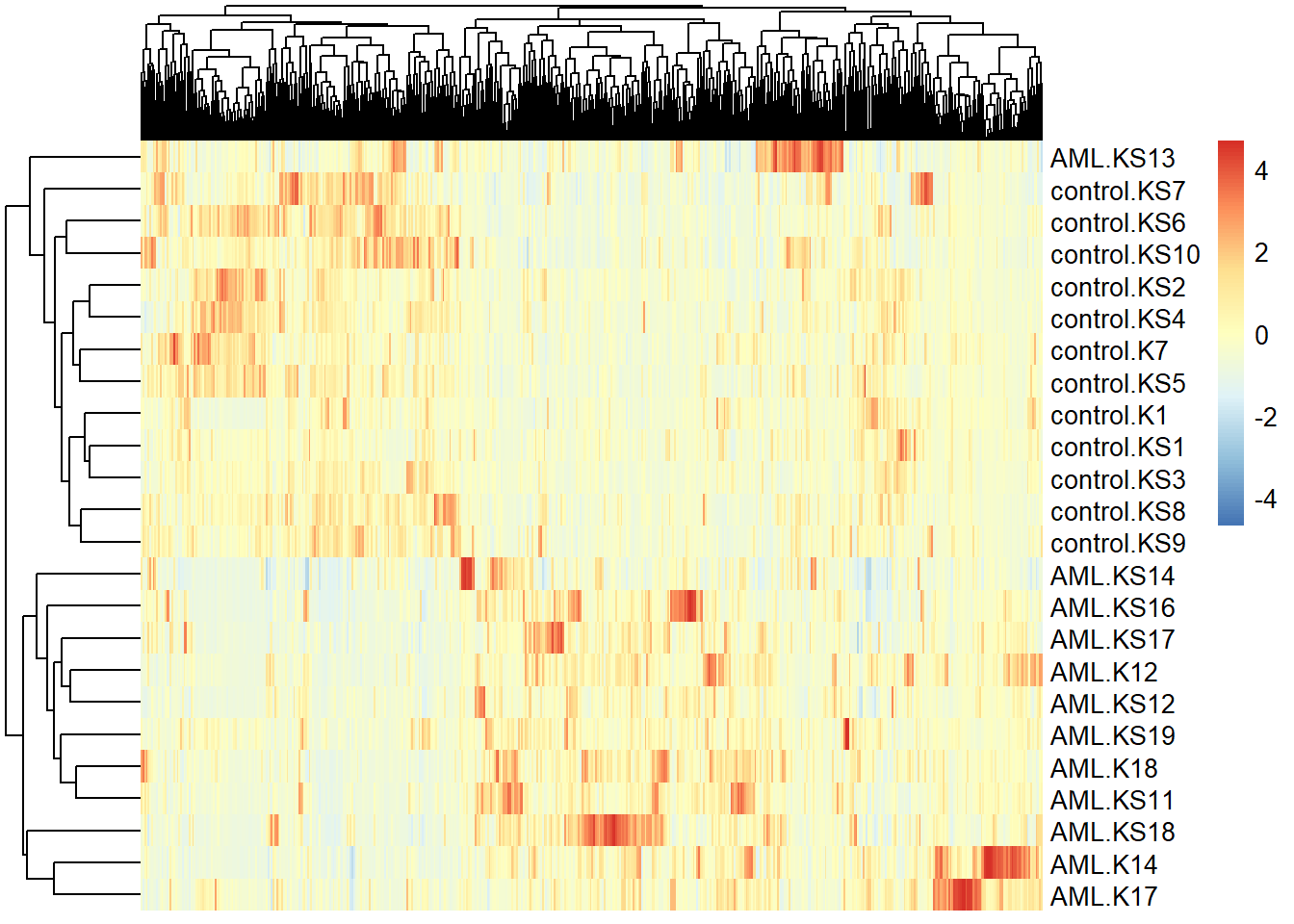
The heatmap seems to suggest that there are definite differences between treated individuals and controls.
3.2.1.1 Performing a little GO-term analysis
GO term analysis is generally conducted after differential expression analysis, but since the link to the data Sean provided did not have a count matrix (nor do I have the means to generate one as I do not have the .fastq or the SAM / BAM files for the data), there was just no way for me to conform to the traditional workflow.
Nevertheless, I thought that it would still be interesting to have a feel for what cellular processes are the most affected as a result of the above heatmap. Hence, I conducted a GO term analysis to see if anything would pop up.
Note that in the below code block, I set organism to hsapiens as we’re dealing with human samples here. All annotations for the genes come from EnsemblGenomes (i.e., “ENSG” - this is the default database that gconvert() will search) - the results of below code block has been saved into a variable called GOresults:
GOresults <- gconvert(colnames(transposed), organism = "hsapiens", target = "ENSG")The contents of GOresults (not including the first three columns) are shown below:
| target | name | description | namespace |
|---|---|---|---|
| ENSG00000198997 | MIR107 | microRNA 107 [Source:HGNC Symbol;Acc:HGNC:31496] | MIRBASE |
| ENSG00000221783 | MIR1183 | microRNA 1183 [Source:HGNC Symbol;Acc:HGNC:35264] | MIRBASE |
| ENSG00000221745 | MIR1197 | microRNA 1197 [Source:HGNC Symbol;Acc:HGNC:35376] | MIRBASE |
| ENSG00000283203 | MIR1246 | microRNA 1246 [Source:HGNC Symbol;Acc:HGNC:35312] | MIRBASE |
| ENSG00000283958 | MIR1248 | microRNA 1248 [Source:HGNC Symbol;Acc:HGNC:35314] | MIRBASE |
| ENSG00000221598 | MIR1249 | microRNA 1249 [Source:HGNC Symbol;Acc:HGNC:35315] | MIRBASE |
| ENSG00000221265 | MIR1255A | microRNA 1255a [Source:HGNC Symbol;Acc:HGNC:35320] | MIRBASE |
| ENSG00000221754 | MIR1260A | microRNA 1260a [Source:HGNC Symbol;Acc:HGNC:35325] | MIRBASE |
| ENSG00000266192 | MIR1260B | microRNA 1260b [Source:HGNC Symbol;Acc:HGNC:38258] | MIRBASE |
| ENSG00000221680 | MIR1278 | microRNA 1278 [Source:HGNC Symbol;Acc:HGNC:35356] | MIRBASE |
| ENSG00000221264 | MIR1284 | microRNA 1284 [Source:HGNC Symbol;Acc:HGNC:35362] | MIRBASE |
| ENSG00000281842 | MIR1291 | microRNA 1291 [Source:HGNC Symbol;Acc:HGNC:35284] | MIRBASE |
| ENSG00000221430 | MIR1294 | microRNA 1294 [Source:HGNC Symbol;Acc:HGNC:35287] | MIRBASE |
| ENSG00000275377 | MIR1299 | microRNA 1299 [Source:HGNC Symbol;Acc:HGNC:35290] | MIRBASE |
| ENSG00000221552 | MIR1303 | microRNA 1303 [Source:HGNC Symbol;Acc:HGNC:35301] | MIRBASE |
| ENSG00000223109 | MIR1538 | microRNA 1538 [Source:HGNC Symbol;Acc:HGNC:35382] | MIRBASE |
| ENSG00000207695 | MIR184 | microRNA 184 [Source:HGNC Symbol;Acc:HGNC:31555] | MIRBASE |
| ENSG00000215938 | MIR190B | microRNA 190b [Source:HGNC Symbol;Acc:HGNC:33656] | MIRBASE |
| ENSG00000238705 | MIR1976 | microRNA 1976 [Source:HGNC Symbol;Acc:HGNC:37064] | MIRBASE |
| ENSG00000207568 | MIR203A | microRNA 203a [Source:HGNC Symbol;Acc:HGNC:31581] | MIRBASE |
| ENSG00000207604 | MIR206 | microRNA 206 [Source:HGNC Symbol;Acc:HGNC:31584] | MIRBASE |
| ENSG00000284442 | MIR2110 | microRNA 2110 [Source:HGNC Symbol;Acc:HGNC:37071] | MIRBASE |
| ENSG00000212102 | MIR301B | microRNA 301b [Source:HGNC Symbol;Acc:HGNC:33667] | MIRBASE |
| ENSG00000265396 | MIR3128 | microRNA 3128 [Source:HGNC Symbol;Acc:HGNC:38188] | MIRBASE |
| ENSG00000264931 | MIR3138 | microRNA 3138 [Source:HGNC Symbol;Acc:HGNC:38341] | MIRBASE |
| ENSG00000264823 | MIR3154 | microRNA 3154 [Source:HGNC Symbol;Acc:HGNC:38176] | MIRBASE |
| ENSG00000264226 | MIR3168 | microRNA 3168 [Source:HGNC Symbol;Acc:HGNC:38249] | MIRBASE |
| ENSG00000199090 | MIR326 | microRNA 326 [Source:HGNC Symbol;Acc:HGNC:31769] | MIRBASE |
| ENSG00000284186 | MIR3615 | microRNA 3615 [Source:HGNC Symbol;Acc:HGNC:38905] | MIRBASE |
| ENSG00000198973 | MIR375 | microRNA 375 [Source:HGNC Symbol;Acc:HGNC:31868] | MIRBASE |
| ENSG00000264803 | MIR378C | microRNA 378c [Source:HGNC Symbol;Acc:HGNC:38374] | MIRBASE |
| ENSG00000263831 | MIR378E | microRNA 378e [Source:HGNC Symbol;Acc:HGNC:41671] | MIRBASE |
| ENSG00000283829 | MIR378I | microRNA 378i [Source:HGNC Symbol;Acc:HGNC:41620] | MIRBASE |
| ENSG00000266320 | MIR3909 | microRNA 3909 [Source:HGNC Symbol;Acc:HGNC:38987] | MIRBASE |
| ENSG00000263885 | MIR3920 | microRNA 3920 [Source:HGNC Symbol;Acc:HGNC:38974] | MIRBASE |
| ENSG00000202566 | MIR421 | microRNA 421 [Source:HGNC Symbol;Acc:HGNC:32793] | MIRBASE |
| ENSG00000199156 | MIR422A | microRNA 422a [Source:HGNC Symbol;Acc:HGNC:31879] | MIRBASE |
| ENSG00000265483 | MIR4443 | microRNA 4443 [Source:HGNC Symbol;Acc:HGNC:41830] | MIRBASE |
| ENSG00000264408 | MIR4470 | microRNA 4470 [Source:HGNC Symbol;Acc:HGNC:41833] | MIRBASE |
| ENSG00000263790 | MIR4473 | microRNA 4473 [Source:HGNC Symbol;Acc:HGNC:41540] | MIRBASE |
| ENSG00000264712 | MIR4504 | microRNA 4504 [Source:HGNC Symbol;Acc:HGNC:41624] | MIRBASE |
| ENSG00000264737 | MIR4511 | microRNA 4511 [Source:HGNC Symbol;Acc:HGNC:41589] | MIRBASE |
| ENSG00000284565 | MIR451A | microRNA 451a [Source:HGNC Symbol;Acc:HGNC:32053] | MIRBASE |
| ENSG00000266140 | MIR4533 | microRNA 4533 [Source:HGNC Symbol;Acc:HGNC:41886] | MIRBASE |
| ENSG00000271898 | MIR4791 | microRNA 4791 [Source:HGNC Symbol;Acc:HGNC:41704] | MIRBASE |
| ENSG00000283736 | MIR484 | microRNA 484 [Source:HGNC Symbol;Acc:HGNC:32341] | MIRBASE |
| ENSG00000283726 | MIR5193 | microRNA 5193 [Source:HGNC Symbol;Acc:HGNC:43534] | MIRBASE |
| ENSG00000212040 | MIR543 | microRNA 543 [Source:HGNC Symbol;Acc:HGNC:33664] | MIRBASE |
| ENSG00000221333 | MIR548K | microRNA 548k [Source:HGNC Symbol;Acc:HGNC:35285] | MIRBASE |
| ENSG00000212017 | MIR548U | microRNA 548u [Source:HGNC Symbol;Acc:HGNC:38316] | MIRBASE |
| ENSG00000266721 | MIR5695 | microRNA 5695 [Source:HGNC Symbol;Acc:HGNC:43548] | MIRBASE |
| ENSG00000207627 | MIR581 | microRNA 581 [Source:HGNC Symbol;Acc:HGNC:32837] | MIRBASE |
| ENSG00000208022 | MIR618 | microRNA 618 [Source:HGNC Symbol;Acc:HGNC:32874] | MIRBASE |
| ENSG00000207631 | MIR641 | microRNA 641 [Source:HGNC Symbol;Acc:HGNC:32897] | MIRBASE |
| ENSG00000211575 | MIR760 | microRNA 760 [Source:HGNC Symbol;Acc:HGNC:33666] | MIRBASE |
| ENSG00000283880 | MIR7704 | microRNA 7704 [Source:HGNC Symbol;Acc:HGNC:50089] | MIRBASE |
| ENSG00000284160 | MIR7706 | microRNA 7706 [Source:HGNC Symbol;Acc:HGNC:50110] | MIRBASE |
| ENSG00000276083 | MIR7976 | microRNA 7976 [Source:HGNC Symbol;Acc:HGNC:50092] | MIRBASE |
3.2.2 Constructing a correlation plot using corrplot()
I made a correlation matrix using the base R function cor() and the corrplot() function from the corrplot package.
I did this because I wanted to know if the data was reproducible - that is, whether or not they varied according to sample and treatment conditions.
In the previous correlation matrix I made, I accidentally used the wrong values - I should have used the samples’ (i.e., patients’) gene expression values instead:
data %>% cor() %>% corrplot(method = "square")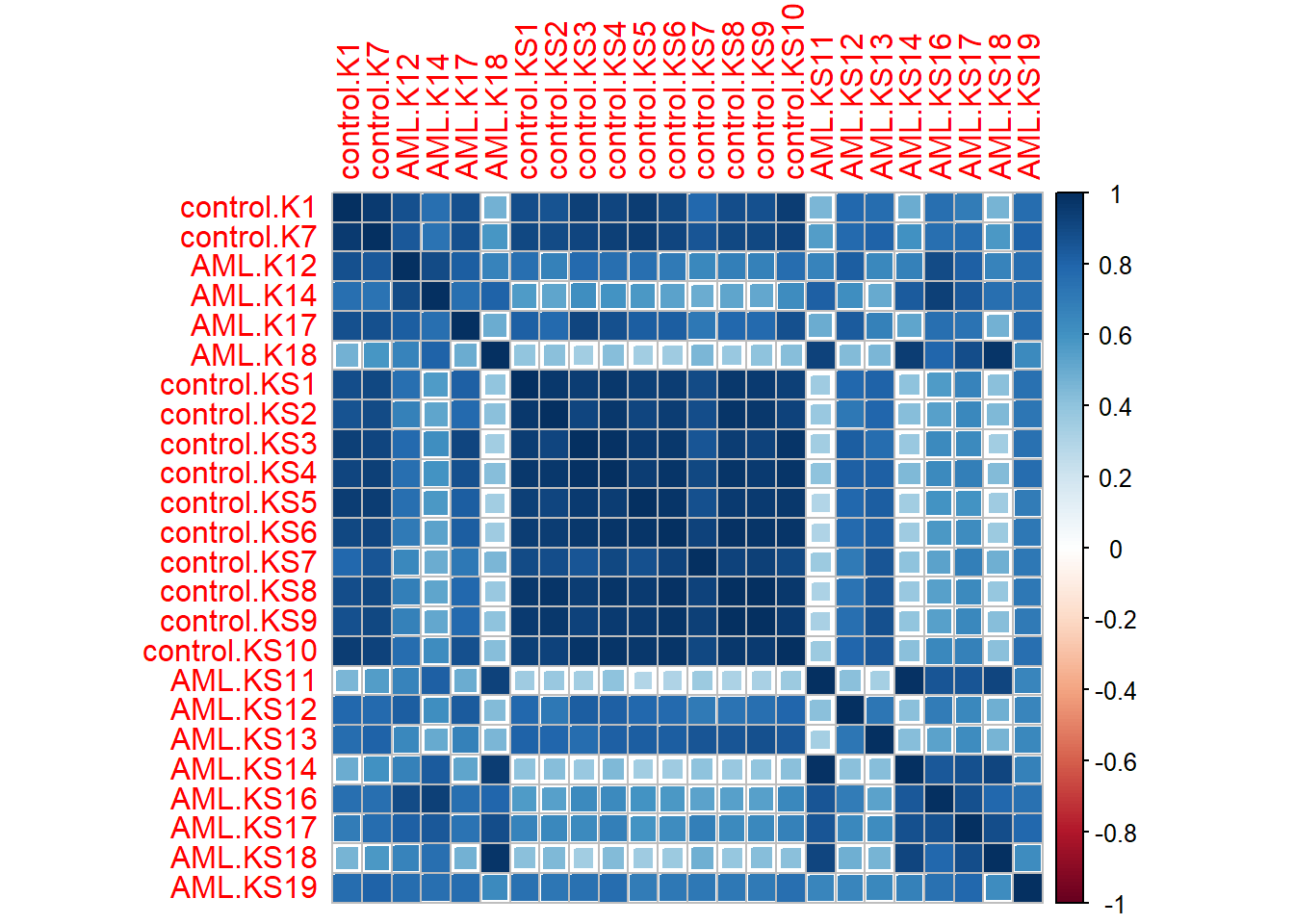
Almost immediately, it was clear that good number of samples are reproducible (i.e., patients whose gene expression values are more or less the same) across different treatment conditions.
3.3 Perfoming PCA
Given the amount of genes in transposed and the above correlation matrix, it was apparent that PCA had to be done.
Training the models using all of the genes in transposed would have taken far too long, not to mention that it is impossible to visualize the gene expression values in 500+ dimensions.
Hence, I performed PCA via the base R function prcomp():
PCAdata <- transposed %>% select(-Status) %>% prcomp(scale. = TRUE)And from this, I made two plots - a PCA plot using ggplot2:
ggplot(as.data.frame(PCAdata$x), aes(x = PCAdata$x[, 1], y = PCAdata$x[, 2])) +
geom_point(aes(col = transposed$Status)) +
labs(title = "PCA Plot of Cancer Data",
x = "PC1",
y = "PC2") + scale_color_discrete(name = "Status")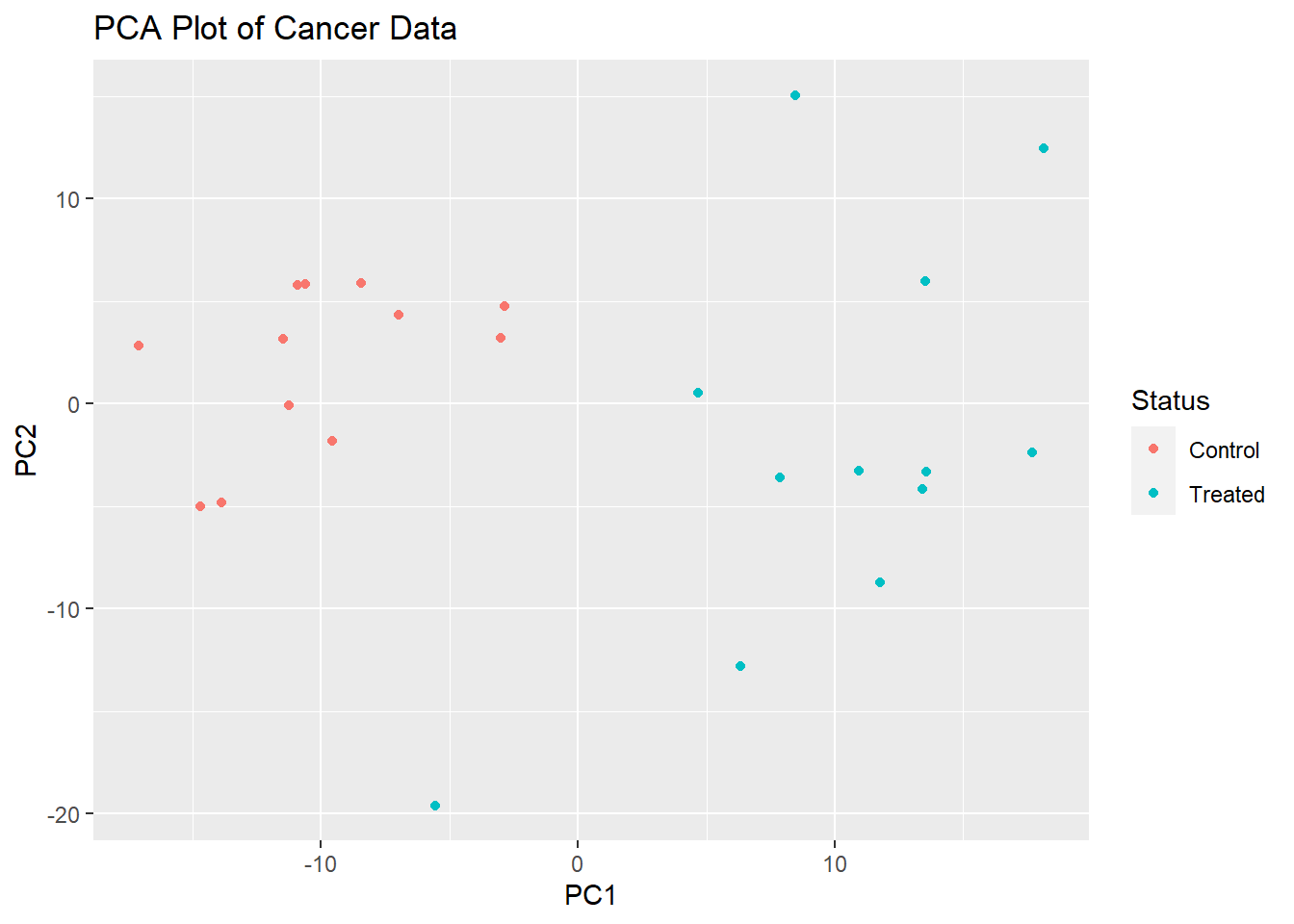
And a screeplot using the base R function barplot():
(PCAdata$x^2 / sum(PCAdata$x^2)) %>% as.data.frame() %>% colSums() %>%
barplot(main = "Screeplot of Transformed Data",
xlab = "Principal Component",
ylab = "Proportion of Explained Variance", las = 1,
col = "slategray2", ylim = c(0, 0.30))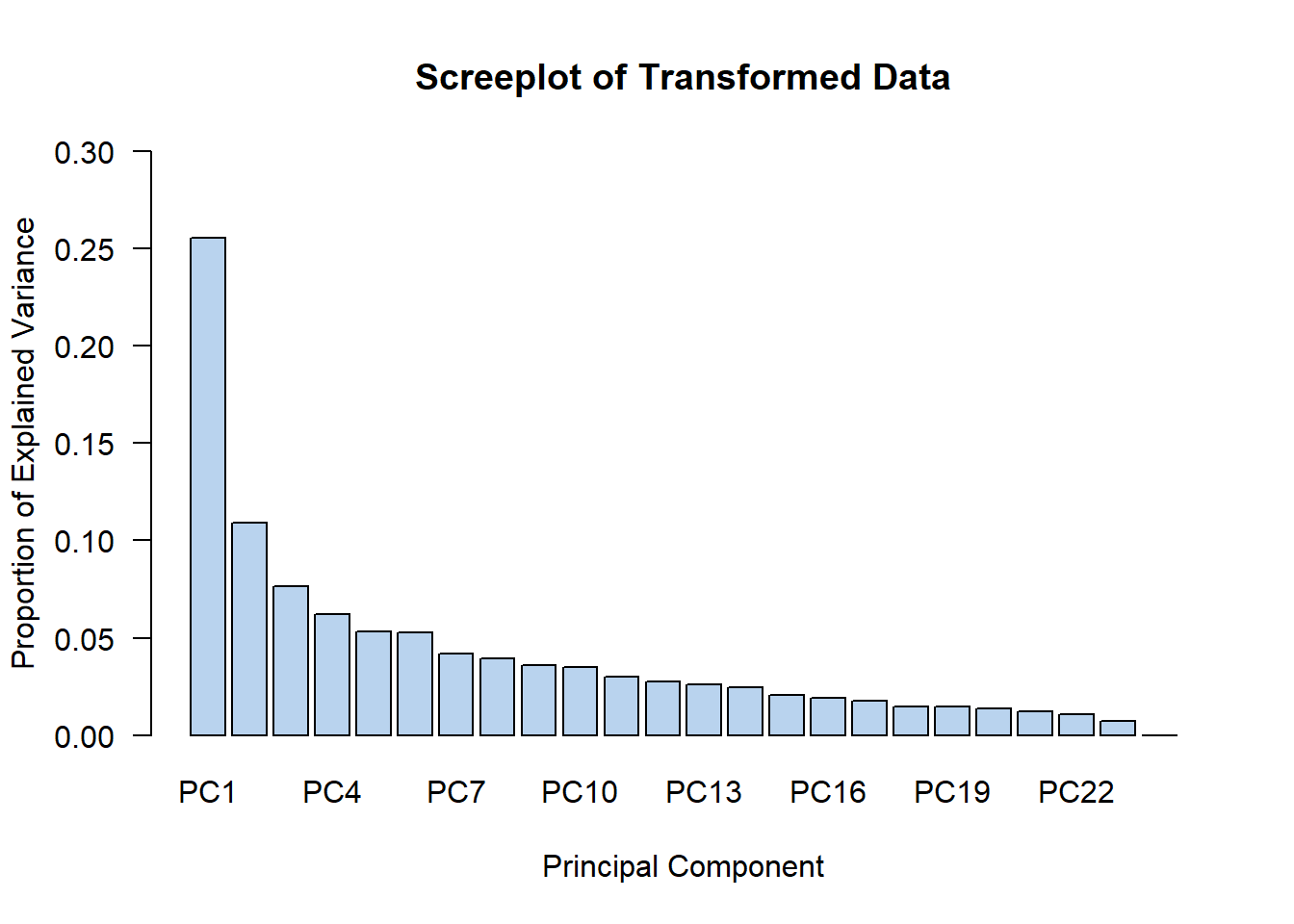
I personally thought that since PC1 and PC2 contained the highest explained variances out of all principal components (this is to be expected anyways), that we could go ahead and PC1 and PC2 to train our machine learning classifiers on.
3.4 Training the Models
3.4.1 Making the training and testing data
For this bit, I took the principal components from PCAdata. I then decided to combine them into a dataframe toUse via the cbind() function.
I also renamed a column using the rename() function from dplyr:
toUse <- cbind(PCAdata$x, transposed$Status) %>% as.data.frame()
toUse$Status <- toUse[, ncol(toUse)] ; toUse[, ncol(toUse)] <- NULL
toUse[, ncol(toUse)] <- as.factor(toUse[, ncol(toUse)])
toUse <- toUse %>% rename(Status = `V25`)I also included the statuses of the patients in toUse.
I then made my training and testing sets via the createDataPartition() function - I also checked the dimensions of my training and testing sets of data using the dim() function:
set.seed(123)
index <- createDataPartition(toUse$Status, p = 0.6, list = FALSE)
training <- toUse[index, ] ; testing <- toUse[-index, ]
dim(training) ; dim(testing)## [1] 16 25## [1] 8 25Note that the set.seed() function was used to ensure reproducibility! The value inside set.seed() can be anything as long as it is numeric.
3.4.2 Performing cross validation
Thereafter, I used the trainControl() function to perform a 10-fold cross validation (as this is the most common value of “k” in k-fold cross validation) - I saved the results of trainControl() in a variable called kfold:
kfold <- trainControl(method = "cv", n = 10)3.4.3 Training the models
I trained several classification models according - the source code for their training are shown for a:
1) kNN algorithm
kNN <- train(Status ~ PC1 + PC2, data = training, method = "knn", trControl = kfold) %>% suppressWarnings()2) linear support vector machine
linearSVM <- tune.svm(Status ~ ., data = training, kernel = "linear")I also managed to construct a visual representation of the support vector machine:
plot(linearSVM$best.model, data = training, PC2 ~ PC1)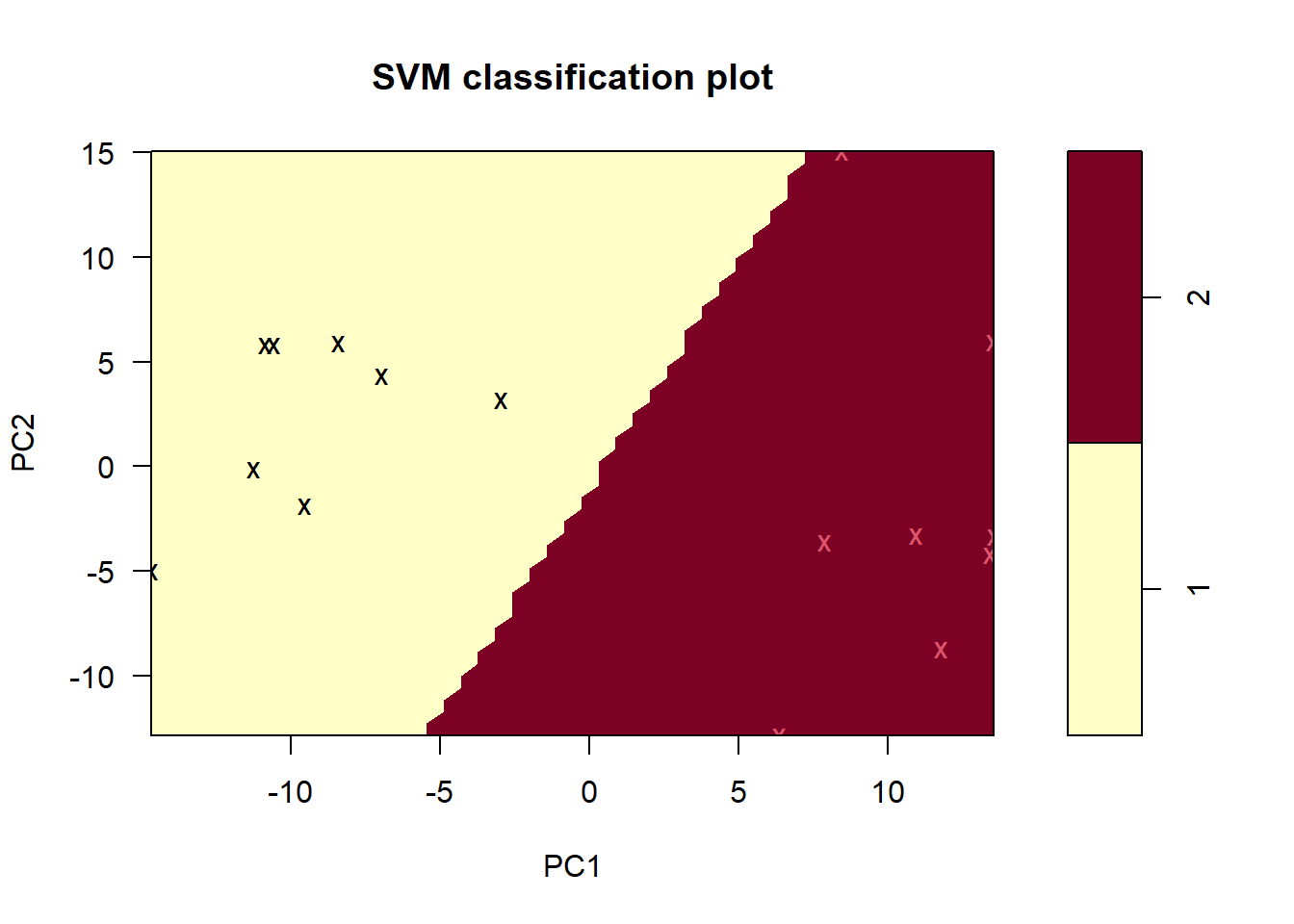
3) Radial support vector machine**
radialSVM <- tune.svm(Status ~ ., data = training, kernel = "radial")And its plot:
plot(radialSVM$best.model, data = training, PC2 ~ PC1)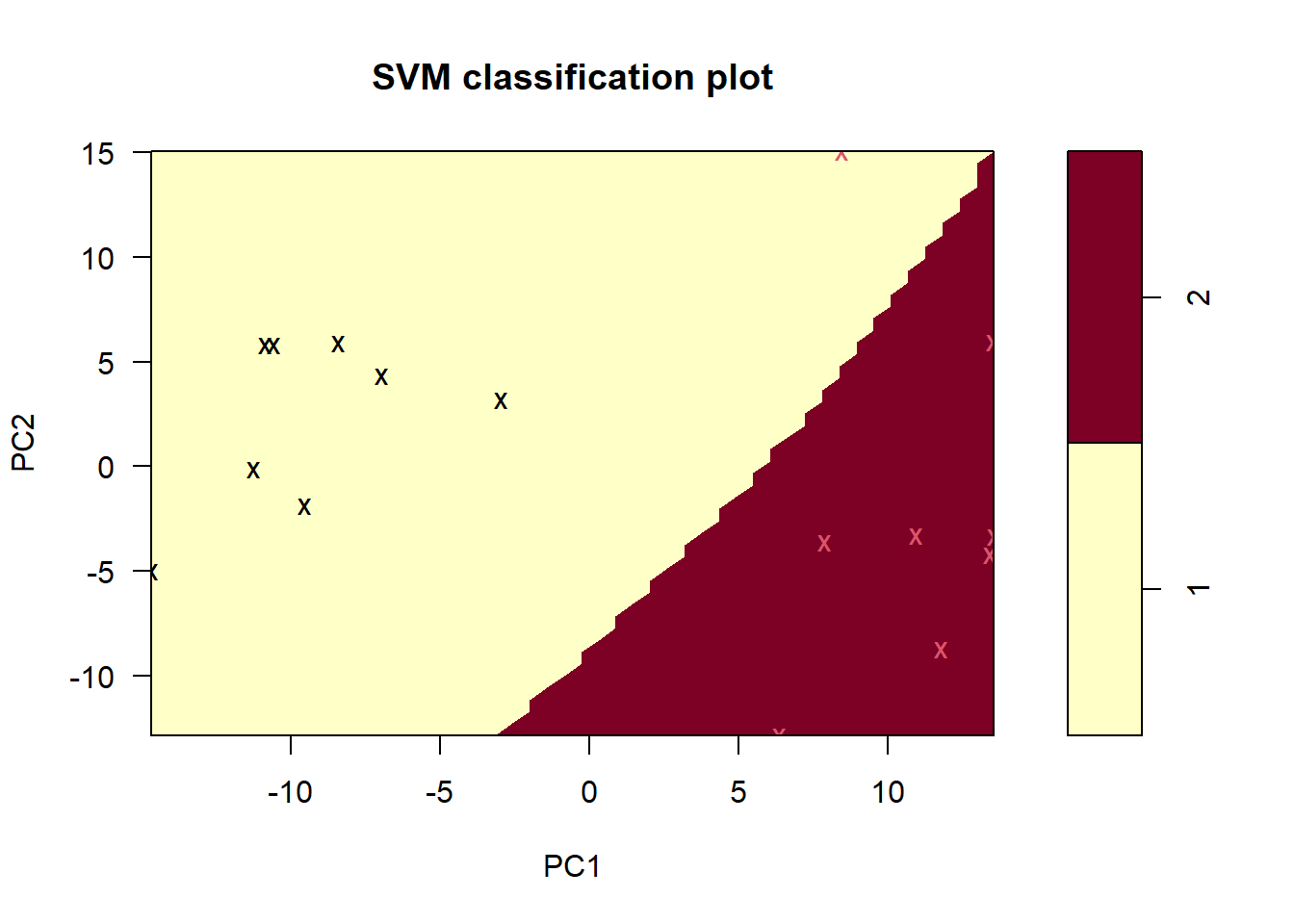
4) Decision tree
rpart <- train(Status ~ ., data = training, method = "rpart",
control = rpart::rpart.control(minsplit = 1),
trControl = kfold) %>% suppressWarnings()I purposely set minsplit to 1 in the control parameter. This is because we have a very small sample size - the minsplit parameter determines how many samples a split in the tree must benefit before the split is introduced as part of the model.
Nevertheless, the decision tree is shown below (NB: “2” are the treated samples):
rattle::fancyRpartPlot(rpart$finalModel)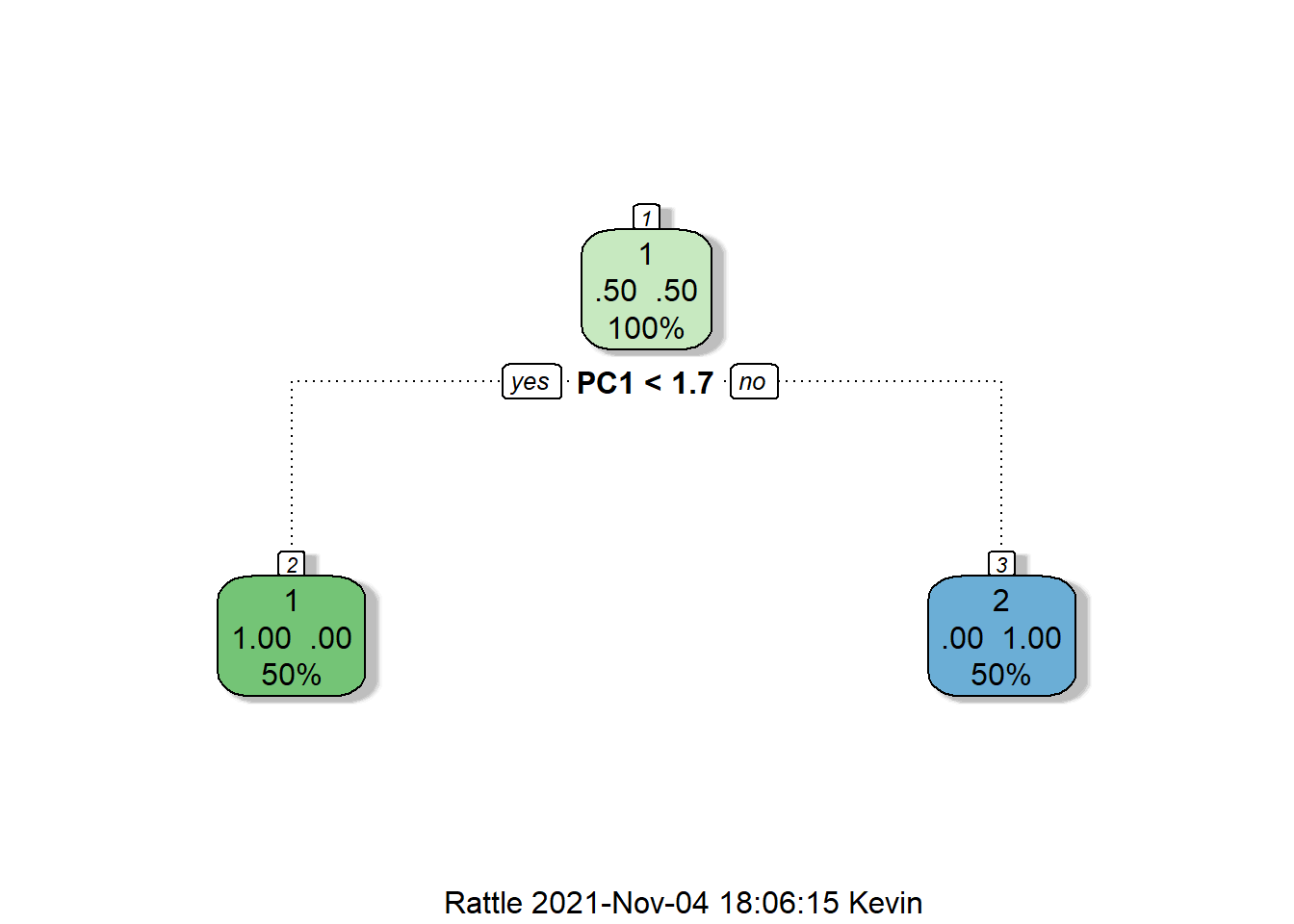
Out of curiousity, I also decided to make a variable importance plot to which principal components actually play a huge role in splitting the tree:
features <- vip(rpart, geom = "point", num_features = 24)
vip(rpart, geom = "point", num_features = 24)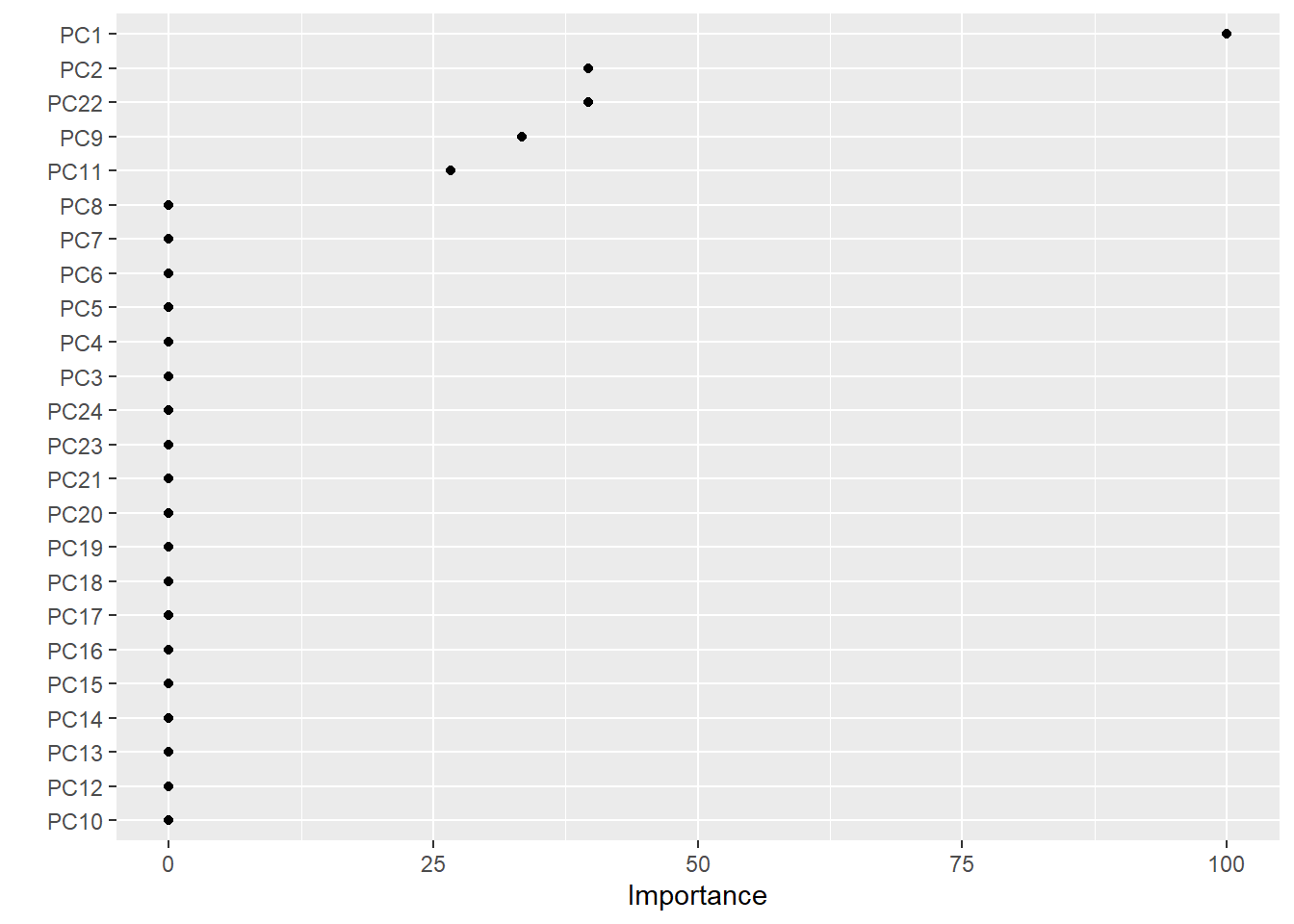
Principal components 1 and 2 were the features that had the highest variable importance. Out of curiosity, I then decided to retrain the model with the principal components that had a variable importance greater than 0:
feat <- features$data %>% filter(Importance > 0) %>% select(Variable) %>% unclass()
feat2 <- feat$Variable %>% paste(collapse = " + ")
rpart <- train(as.formula(paste0("Status ~ ", feat2)), data = training, method = "rpart",
control = rpart::rpart.control(minsplit = 1),
trControl = kfold) %>% suppressWarnings()And the decision tree looks like this:
rattle::fancyRpartPlot(rpart$finalModel)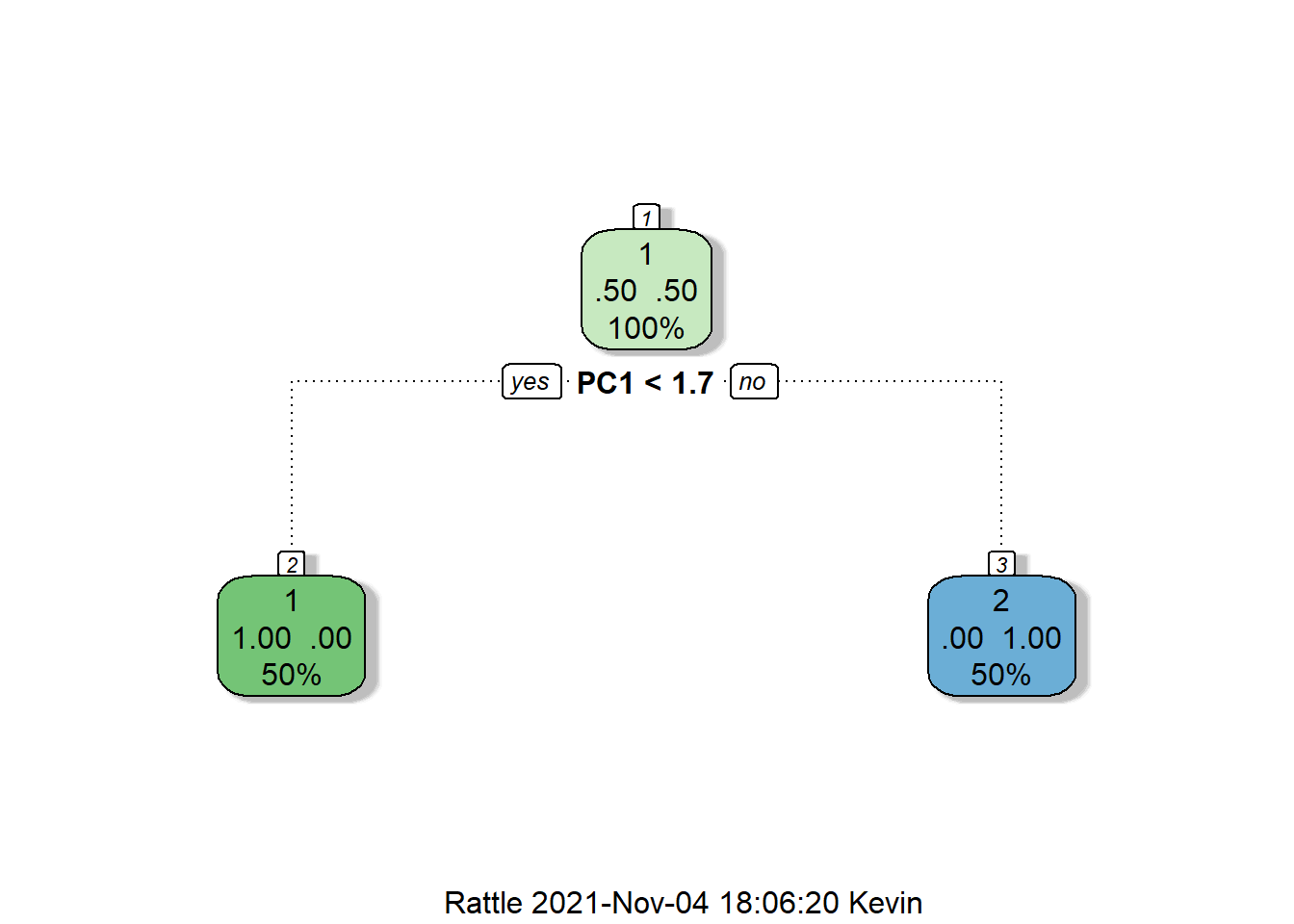
5) Ranger random forest
This is a supposedly “fast” variant of the original random forest algorithm:
ranger <- train(Status ~ ., data = training, method = "ranger", trControl = kfold) %>% suppressWarnings()3.5 Evaluating the Models
I used the confusionMatrix() function from caret to generate a confusion matrix and from it, calculate the sensitivity (i.e., precision) and the recall of the model.
I also used the ROCR package to create a function plotROC() to not only plot the ROC curve of a model, but also calculate the AUC from the ROC curve:
plotROC <- function(model, testing, title) {
AUCval <- predict(model, testing) %>% as.numeric() %>%
prediction(as.numeric(testing$Status)) %>%
performance(measure = "auc")
AUCval <- AUCval@y.values[[1]]
# Make the plot
predict(model, testing) %>% as.numeric() %>%
prediction(as.numeric(testing$Status)) %>%
performance("tpr", "fpr") %>% plot(main = paste0("ROC Curve of ", title),
las = 1,
colorize = TRUE)
grid(lty = "dashed", lwd = 1.5) ; text(paste0("AUC = ", AUCval),
x = 0.7, y = 0.3)
}The performance() function is used to evaluate the model’s predictions against the actual results. The prediction() function is used to create an object of class prediction - while I am unsure why, this step is neccesary to generate ROC plots with ROCR!
Nevertheless, the models and their evaluations are:
1) kNN
kNNstats <- confusionMatrix(predict(kNN, testing), testing$Status)
kNNstats$byClass## Sensitivity Specificity Pos Pred Value
## 1.0000000 0.7500000 0.8000000
## Neg Pred Value Precision Recall
## 1.0000000 0.8000000 1.0000000
## F1 Prevalence Detection Rate
## 0.8888889 0.5000000 0.5000000
## Detection Prevalence Balanced Accuracy
## 0.6250000 0.8750000The kNN’s confusion matrix is as such:
kNNstats$table## Reference
## Prediction 1 2
## 1 4 1
## 2 0 3Also note the ROC curve and AUC:
plotROC(kNN, testing, "kNN")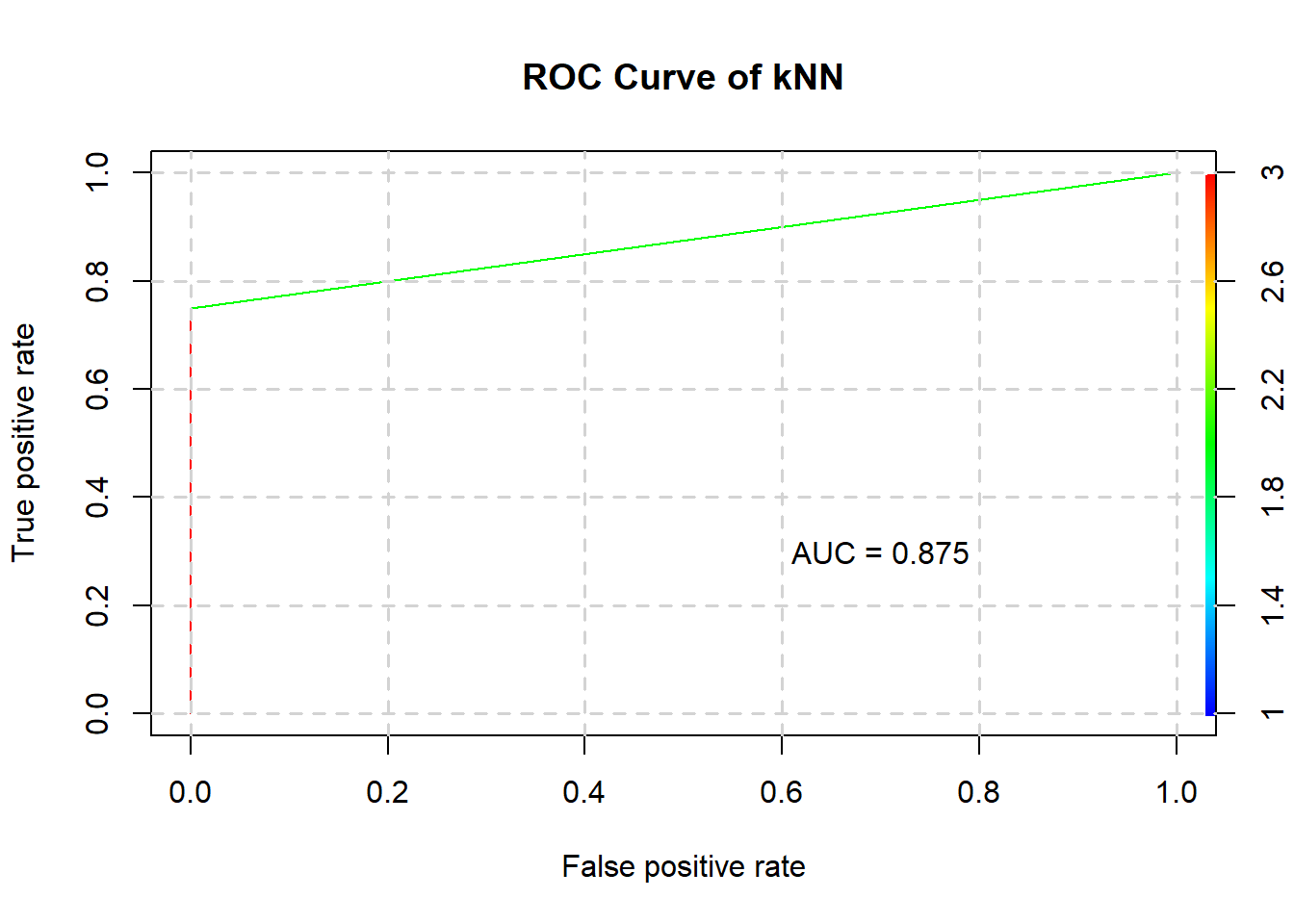
2) Linear support vector machine
linStats <- confusionMatrix(predict(linearSVM$best.model, testing), testing$Status)
linStats$byClass## Sensitivity Specificity Pos Pred Value
## 1.0000000 0.5000000 0.6666667
## Neg Pred Value Precision Recall
## 1.0000000 0.6666667 1.0000000
## F1 Prevalence Detection Rate
## 0.8000000 0.5000000 0.5000000
## Detection Prevalence Balanced Accuracy
## 0.7500000 0.7500000The linear SVM’s confusion matrix is as such:
linStats$table## Reference
## Prediction 1 2
## 1 4 2
## 2 0 2And its ROC curve and AUC:
plotROC(linearSVM$best.model, testing, "Linear SVM")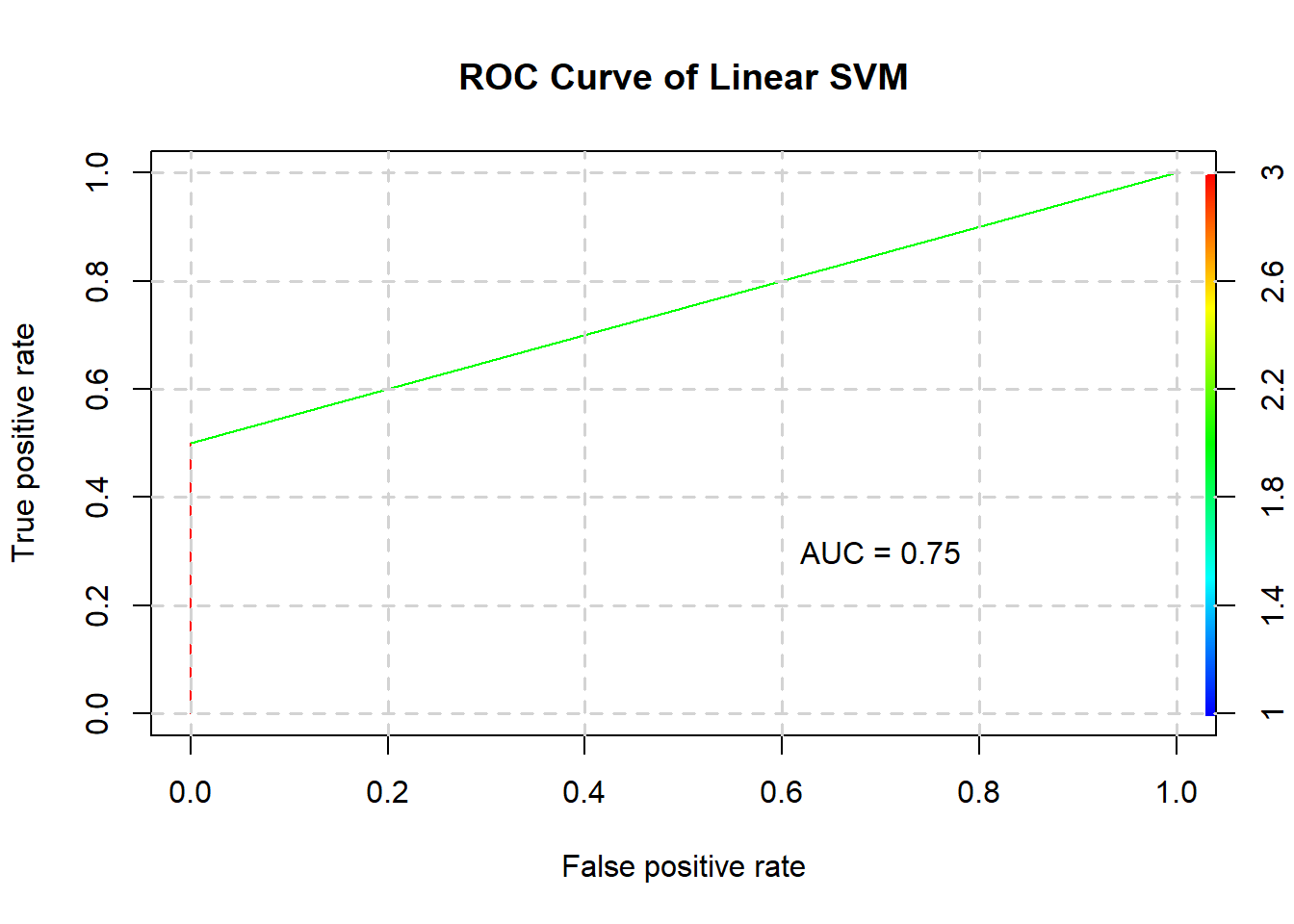
3) Radial support vector machine
radialstats <- confusionMatrix(predict(radialSVM$best.model, testing), testing$Status)
radialstats$byClass## Sensitivity Specificity Pos Pred Value
## 0.7500000 1.0000000 1.0000000
## Neg Pred Value Precision Recall
## 0.8000000 1.0000000 0.7500000
## F1 Prevalence Detection Rate
## 0.8571429 0.5000000 0.3750000
## Detection Prevalence Balanced Accuracy
## 0.3750000 0.8750000The radial SVM’s confusion matrix is as such:
radialstats$table## Reference
## Prediction 1 2
## 1 3 0
## 2 1 4And its ROC curve and AUC:
plotROC(radialSVM$best.model, testing, "Radial SVM")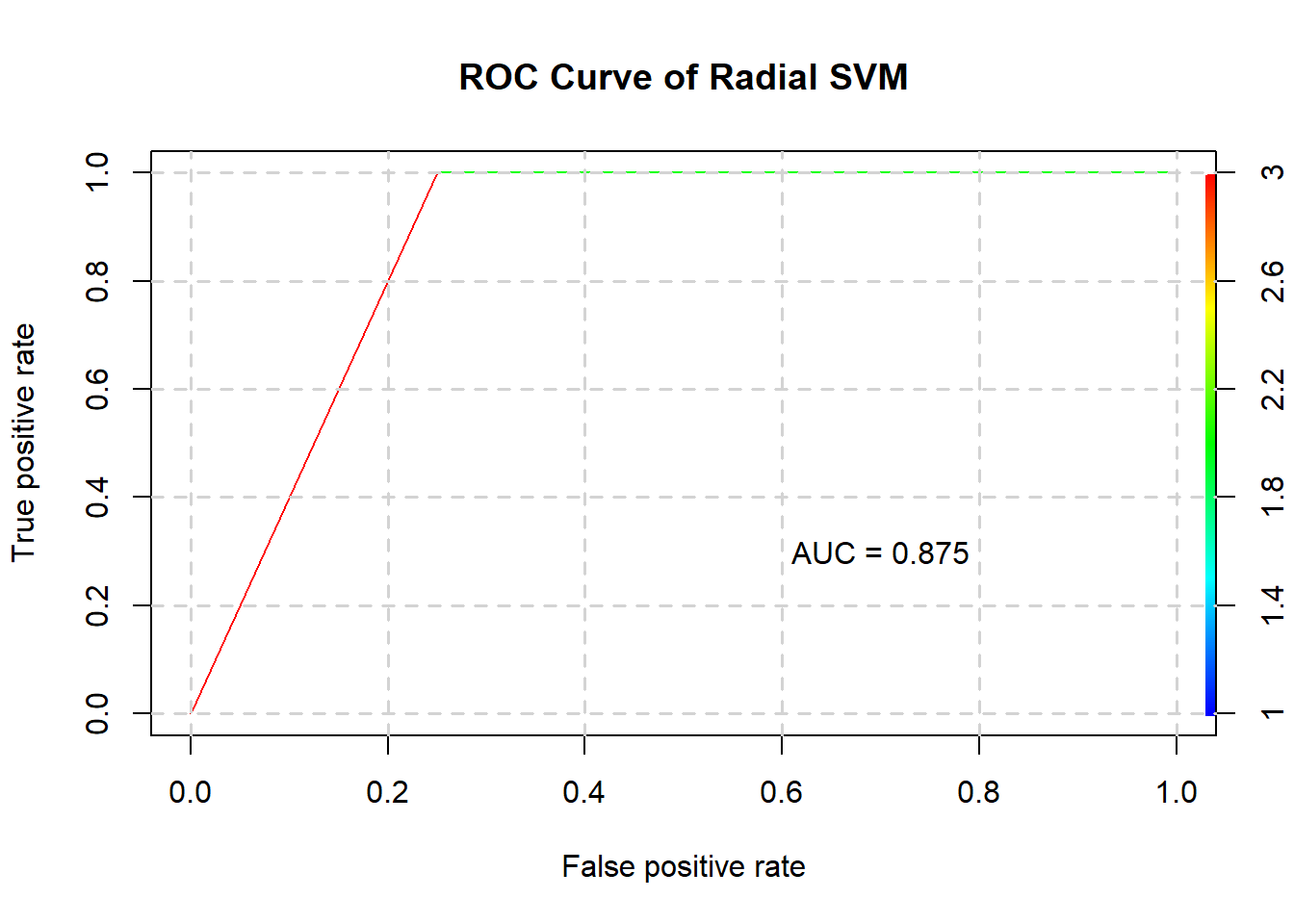
4) Decision tree
rpartstats <- confusionMatrix(predict(rpart, testing), testing$Status)
rpartstats$byClass## Sensitivity Specificity Pos Pred Value
## 1.0000000 0.7500000 0.8000000
## Neg Pred Value Precision Recall
## 1.0000000 0.8000000 1.0000000
## F1 Prevalence Detection Rate
## 0.8888889 0.5000000 0.5000000
## Detection Prevalence Balanced Accuracy
## 0.6250000 0.8750000The decision tree’s confusion matrix is as such:
rpartstats$table## Reference
## Prediction 1 2
## 1 4 1
## 2 0 3And its ROC curve and AUC:
plotROC(rpart, testing, "Decision Tree")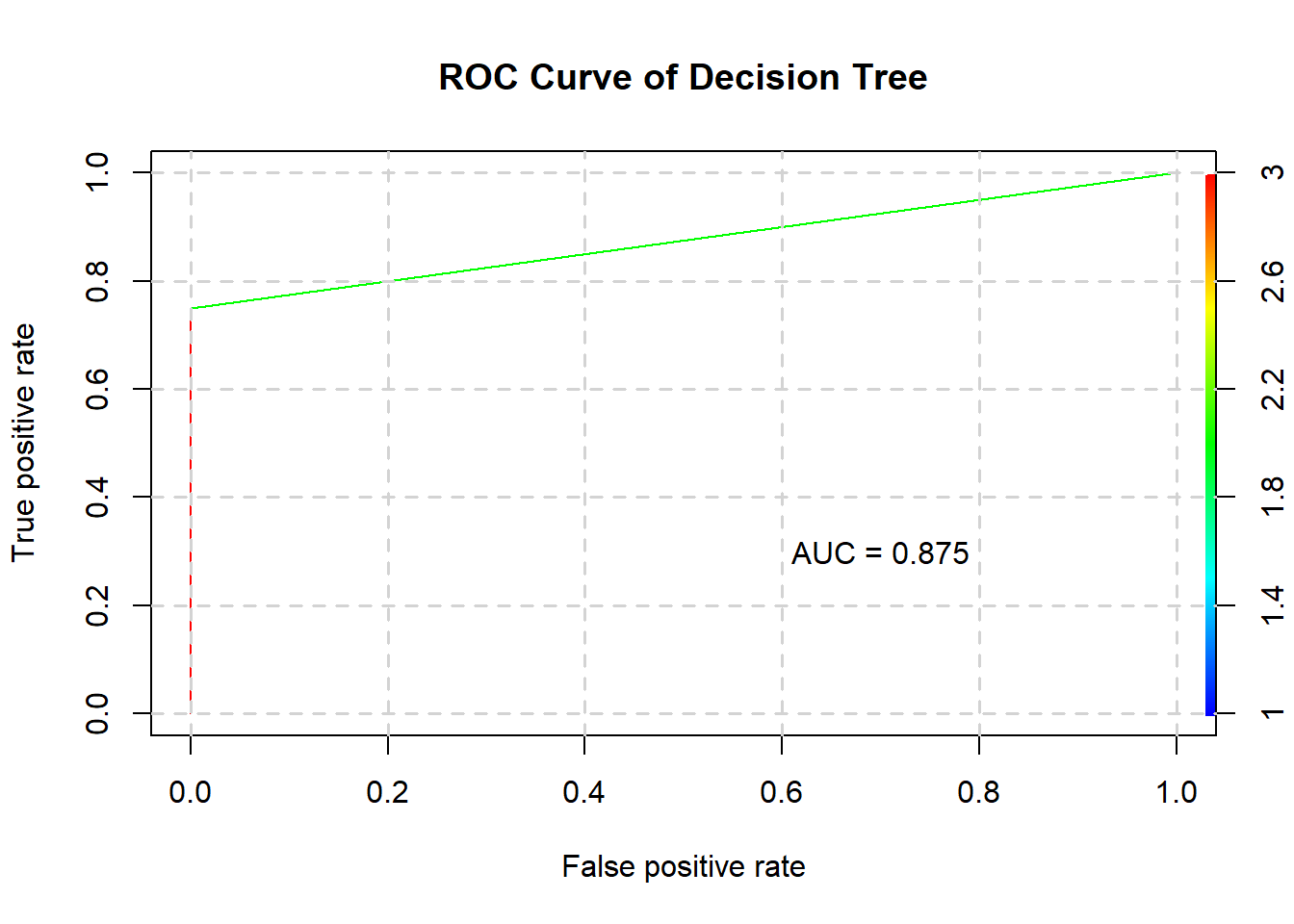
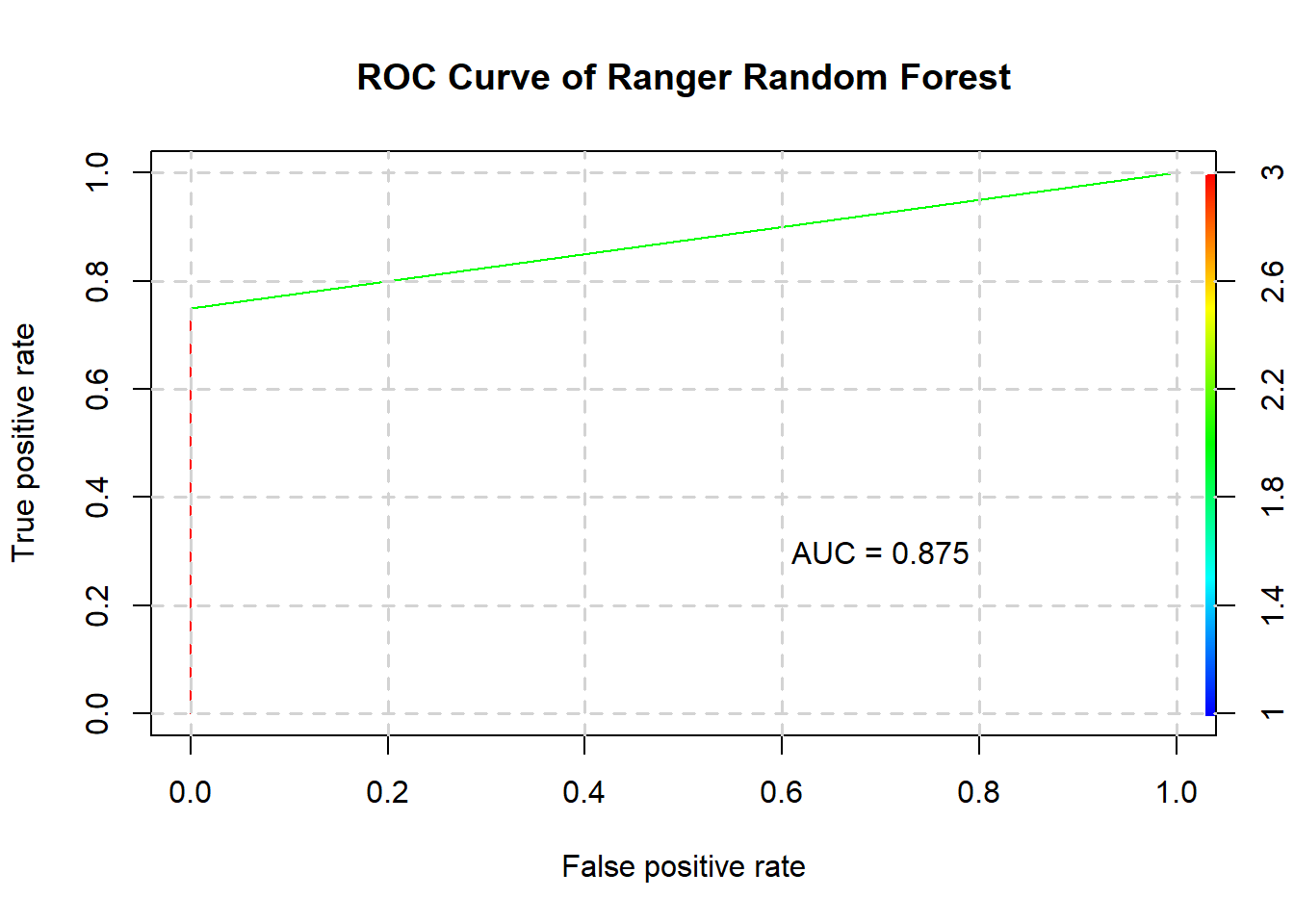
5) Ranger random forest
rangerstats <- confusionMatrix(predict(ranger, testing), testing$Status)
rangerstats$byClass## Sensitivity Specificity Pos Pred Value
## 1.0000000 0.7500000 0.8000000
## Neg Pred Value Precision Recall
## 1.0000000 0.8000000 1.0000000
## F1 Prevalence Detection Rate
## 0.8888889 0.5000000 0.5000000
## Detection Prevalence Balanced Accuracy
## 0.6250000 0.8750000The ranger random forest’s confusion matrix is as such:
rangerstats$table## Reference
## Prediction 1 2
## 1 4 1
## 2 0 3And its ROC curve and AUC:
plotROC(ranger, testing, "Ranger Random Forest")