2 NGS Pipeline: Saccharomyces cerevisiae samples
What follows is a reiteration of the files you should have already downloaded. So hopefully you have your files already in your FASTQ in RNA-genomics directory.
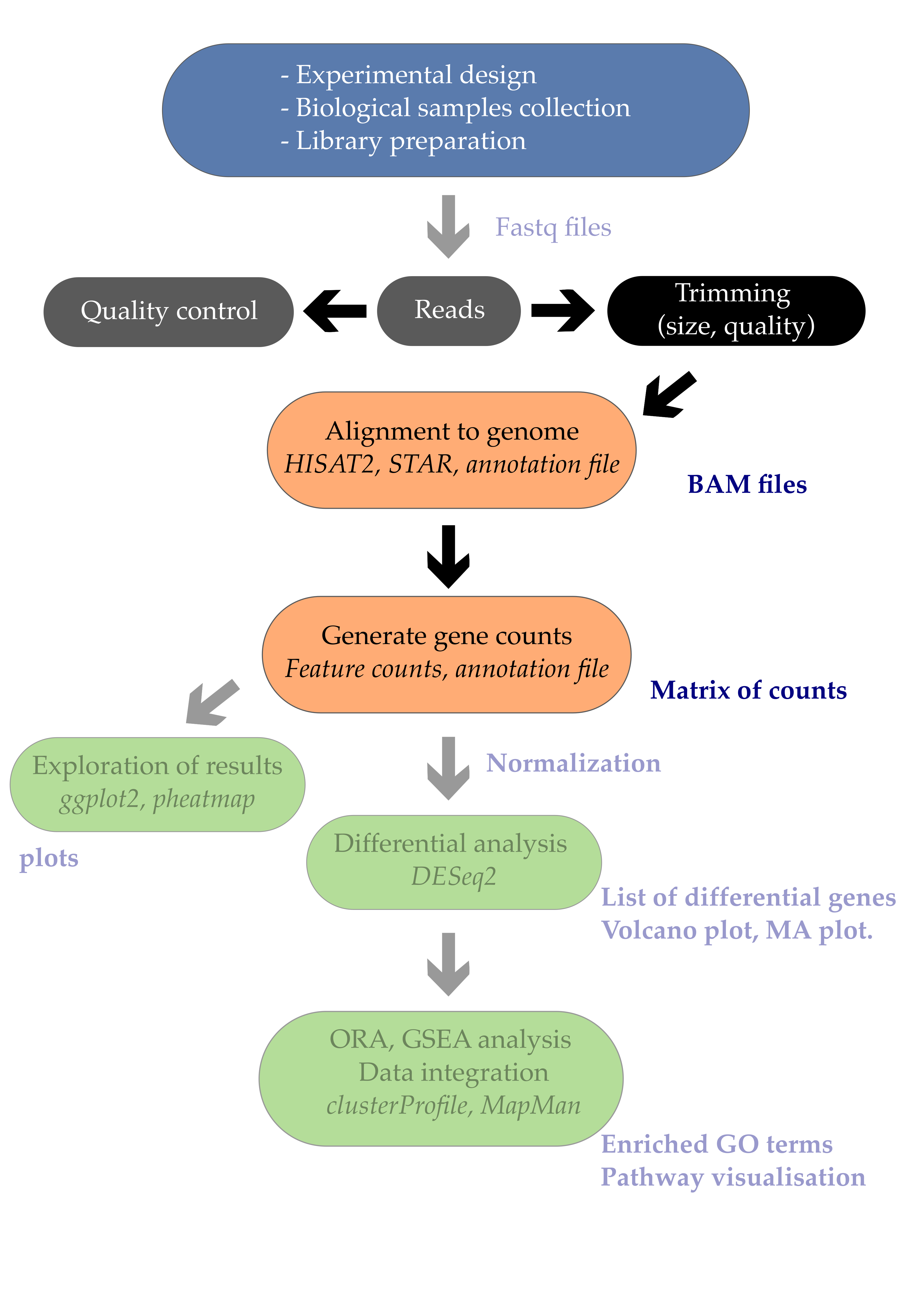
We are going to walk steps of a Next Generation Sequencing pipeline using samples from Saccharomyces cerevisiae because the files are nice and small. Both in R and in the terminal. Like we did with the variant analysis, RNA-seq starts with QC and specifically FASTQC. So since we already walked through that last time we are going to skip over this bit. Navigate to your FASTQ directory:
pwd
cd ~
pwd
cd ~/Documents/RProjects/genomics/RNA-genomics/FASTQ## /Users/ggiaever/Documents/RProjects/genomics/RNA-genomics
## /Users/ggiaeverFor our examples we will focus on RNA-seq as it is the most manageable computationally as opposed to methylation, Chip-chip and DNA variant analysis. Its also, probably for that reason, one of the more frequently used technologies in research, likely for this reasons.
The samples can be downloaded from the ENA: The European Nucleotide Archive (EMBL-EBI)
For most reads presented by ENA, there are three kinds of file available:
Submitted files are identical to those submitted by the user 1. ENA submitted files are available in the ‘run’ directory 2. FASTQ files are archive-generated files generated according to a standardised format (learn more about this format) 3. SRA files are in a format designed to work with NCBI’s SRA Toolkit
Each of the three file types has its own directory on the FTP server. A folder exists for every run, which is named with the accession of the run, e.g. files for run ERR164407 are in a directory named ERR164407. These run accession directories are organised into parent directories named with their first 6 characters. For ERR164407 this is ‘ERR164’. This structure is repeated across all three file types, e.g.
ftp://ftp.sra.ebi.ac.uk/vol1/
It follows that the ftp addresses for our files are as listed below. We can download the files using the curl command. Options: -L, follows redirects, -O, write the output to a file named as the remote file, -R, time the download. First navigate to the FASTQ directory and then type:
cd ~/Documents/RProjects/genomics/RNA-genomics/FASTQ
curl -L -R -O ftp.sra.ebi.ac.uk/vol1/fastq/ERR458/ERR458493/ERR458493.fastq.gz
curl -L -R -O ftp.sra.ebi.ac.uk/vol1/fastq/ERR458/ERR458494/ERR458494.fastq.gz
curl -L -R -O ftp.sra.ebi.ac.uk/vol1/fastq/ERR458/ERR458495/ERR458495.fastq.gz
curl -L -R -O ftp.sra.ebi.ac.uk/vol1/fastq/ERR458/ERR458500/ERR458500.fastq.gz
curl -L -R -O ftp.sra.ebi.ac.uk/vol1/fastq/ERR458/ERR458501/ERR458501.fastq.gz
curl -L -R -O ftp.sra.ebi.ac.uk/vol1/fastq/ERR458/ERR458502/ERR458502.fastq.gzIf you are on a PC, you need to use the wget command. For wget it would be, e.g.:
cd ~/Documents/RProjects/genomics/RNA-genomics/FASTQ
wget ftp.sra.ebi.ac.uk/vol1/fastq/ERR458/ERR458493/ERR458493.fastq.gz
wget ftp.sra.ebi.ac.uk/vol1/fastq/ERR458/ERR458494/ERR458494.fastq.gz
wget ftp.sra.ebi.ac.uk/vol1/fastq/ERR458/ERR458495/ERR458495.fastq.gz
wget ftp.sra.ebi.ac.uk/vol1/fastq/ERR458/ERR458500/ERR458500.fastq.gz
wget ftp.sra.ebi.ac.uk/vol1/fastq/ERR458/ERR458501/ERR458501.fastq.gz
wget ftp.sra.ebi.ac.uk/vol1/fastq/ERR458/ERR458502/ERR458502.fastq.gz
ftp.sra.ebi.ac.uk/vol1/fastq/ERR458/ERR458493/ERR458493.fastq.gz
wget ftp.sra.ebi.ac.uk/vol1/fastq/ERR458/ERR458494/ERR458494.fastq.gz
wget ftp.sra.ebi.ac.uk/vol1/fastq/ERR458/ERR458495/ERR458495.fastq.gz
wget ftp.sra.ebi.ac.uk/vol1/fastq/ERR458/ERR458500/ERR458500.fastq.gz
wget ftp.sra.ebi.ac.uk/vol1/fastq/ERR458/ERR458501/ERR458501.fastq.gz
wget ftp.sra.ebi.ac.uk/vol1/fastq/ERR458/ERR458502/ERR458502.fastq.gzThese files take about 7’ to download.
The two other big files that you have are, first the DNA sequence, and second the GTF file, which is a file describing genomic features. You can get these from the ensembl database, which is the go to database for annotated genomes.
macOS
mkdir -p ~/Documents/RProjects/genomics/RNA-genomics/FASTQ/r64
cd ~/Documents/RProjects/genomics/RNA-genomics/FASTQ/r64
curl -L -R -O ftp://ftp.ensembl.org/pub/release-96/fasta/saccharomyces_cerevisiae/dna/Saccharomyces_cerevisiae.R64-1-1.dna.toplevel.fa.gz
curl -L -R -O ftp://ftp.ensembl.org/pub/release-96/gtf/saccharomyces_cerevisiae/Saccharomyces_cerevisiae.R64-1-1.96.gtf.gzWindows
mkdir -p ~/Documents/RProjects/genomics/RNA-genomics/FASTQ/r64
cd ~/Documents/RProjects/genomics/RNA-genomics/FASTQ/r64
wget ftp://ftp.ensembl.org/pub/release-96/fasta/saccharomyces_cerevisiae/dna/Saccharomyces_cerevisiae.R64-1-1.dna.toplevel.fa.gz
wget ftp://ftp.ensembl.org/pub/release-96/gtf/saccharomyces_cerevisiae/Saccharomyces_cerevisiae.R64-1-1.96.gtf.gz2.1 Fastq files
We’ve talked about and looked at fastq files a little bit, but let’s look at them here in there raw format. First we have to unzip one.
To unzip you use gunzip and you need the -c to leave the gz file behind:
cd ~/Documents/RProjects/genomics/RNA-genomics
gunzip -c FASTQ/ERR458493.fastq.gz > FASTQ/ERR458493.fastq
head -n 4 FASTQ/ERR458493.fastq## @ERR458493.1 DHKW5DQ1:219:D0PT7ACXX:1:1101:1724:2080/1
## CGCAAGACAAGGCCCAAACGAGAGATTGAGCCCAATCGGCAGTGTAGTGAA
## +
## B@@FFFFFHHHGHJJJJJJIJJGIGIIIGI9DGGIIIEIGIIFHHGGHJIBFirst line: A sequence identifier with information about the sequencing run and the cluster. The exact contents of this line vary by based on the BCL to FASTQ conversion software used.
Second line: The sequence (the base calls; A, C, T, G and N).
Third line: A separator, which is simply a plus (+) sign.
Forth line: The base call quality scores. These are Phred +33 encoded, using ASCII characters to represent the numerical quality scores.
We can count the number of lines
cd ~/Documents/RProjects/genomics/RNA-genomics
expr $(cat FASTQ/ERR458493.fastq | wc -l) / 4## 1093957Quality control with fastqc
Use fastqc to check the quality of our fastq files, let’s just look at one quickly since we’ve already looked at one in detail:
cd ~/Documents/RProjects/genomics/RNA-genomics
source $HOME/miniconda3/bin/activate
conda activate rnaseq
fastqc FASTQ/ERR458493.fastq.gz -t 4 -o FASTQC## Started analysis of ERR458493.fastq.gz
## Approx 5% complete for ERR458493.fastq.gz
## Approx 10% complete for ERR458493.fastq.gz
## Approx 15% complete for ERR458493.fastq.gz
## Approx 20% complete for ERR458493.fastq.gz
## Approx 25% complete for ERR458493.fastq.gz
## Approx 30% complete for ERR458493.fastq.gz
## Approx 35% complete for ERR458493.fastq.gz
## Approx 40% complete for ERR458493.fastq.gz
## Approx 45% complete for ERR458493.fastq.gz
## Approx 50% complete for ERR458493.fastq.gz
## Approx 55% complete for ERR458493.fastq.gz
## Approx 60% complete for ERR458493.fastq.gz
## Approx 65% complete for ERR458493.fastq.gz
## Approx 70% complete for ERR458493.fastq.gz
## Approx 75% complete for ERR458493.fastq.gz
## Approx 80% complete for ERR458493.fastq.gz
## Approx 85% complete for ERR458493.fastq.gz
## Approx 90% complete for ERR458493.fastq.gz
## Approx 95% complete for ERR458493.fastq.gz
## Analysis complete for ERR458493.fastq.gzWe’ve already discussed fastqc files so let’s have a look at them.
Adapter sequences: if the data are used for variant analyses, genome annotation or genome or transcriptome assembly purposes, we recommend read trimming, including both, adapter and quality trimming.
• Illumina Universal Adapter—AGATCGGAAGAG • Illumina Small RNA 3’ Adapter—TGGAATTCTCGG • Illumina Small RNA 5’ Adapter—GATCGTCGGACT • Nextera Transposase Sequence—CTGTCTCTTATA
Should I trim adapters from my Illumina reads? This depends on the objective of your experiments. In case you are sequencing for counting applications like differential gene expression (DGE) RNA-seq analysis, ChIP-seq, ATAC-seq, read trimming is generally not required anymore when using modern aligners. For such studies local aligners or pseudo-aligners should be used. Modern “local aligners” like STAR, BWA-MEM, HISAT2, will “soft-clip” non-matching sequences. Pseudo-aligners like Kallisto or Salmon will also not have any problem with reads containing adapter sequences.However, if the data are used for variant analyses, genome annotation or genome or transcriptome assembly purposes, we recommend read trimming, including both, adapter and quality trimming.
We will therefore skip trimming our reads and we will use Subread to align our reads.
Aligning is just a simple command in the terminal. First we need to build an index using the reference fasta DNA sequence. Why do we need an index? Genomes can be very large. A genome index allows the aligner to rapidly look up candidate alignment locations for each read rather than conduct an exhaustive search each time.
Most aligners need to create their own indexes, so here we will use the subread-buildindex command module to make an index file for the yeast genome. It will create a set of files, these files together comprise the index. The commands to generate the index looks like this:
./subread-align [options] -i2.2 Mandatory arguments:
-i
-r
-t