3 Data
3.1 Outline
- Section 3.2 lists and loads R packages used in Part 1 of this book
- Section 3.3 introduces and reads in all raw data files
- Section 3.4 provides a data dictionary and summary of each variable in each dataset
- Section 3.5 details the data preparation process, and presents code and procedures that turn raw data into files that are ready for analysis
3.2 R Packages
Each time a new R session is initialized, all required packages have to be loaded using library (except the base, stats, and utils packages).
library(AlphaSimR) # Genetic population simulation package
library(bookdown) # Compilation of book
library(dataMeta) # Data dictionaries
library(data.table) # Read in delimited files
library(GALLO) # QTL Annotation/Enrichment
library(gprofiler2) # Gene Annotation/Enrichment
library(Hmisc) # Miscellaneous
library(kableExtra) # Results tables
library(knitr) # RMarkdown report generation
library(lubridate) # Management of date and time variables
library(optiSel) # Evaluation of pedigree file
library(pedigree) # Calculation of generation number
library(qvalue) # Estimation of Q-values
library(tidyverse) # Data management, cleaning, and visualization
library(vroom) # Efficient loading of large files3.3 Files
The following 4 files were communicated from The Maschhoff’s LLC by Dr. Crum:
- Full_Pedigree.csv (40.98 MB; R object name: ped)
- PigInformation.csv (128.33 MB; R object name: pig_info)
- genotypes.txt (6.48 GB; R object name: genotype)
- 50K_Porcine_n49991_header_20190528.map (1.32 MB; R object name: map)
Based on the size of these files, a simple workaround using an outside R script was conducted to reduce time spent loading each file.
# readdata.r script described below (this script is not evaluated in this document)
rm(list = ls())
### Change working directory
setwd("C:/Users/cgroh/OneDrive/Desktop/PhD/Project 1 - GSM/Raw Data/")
### Read in 4 files
genotype <- fread('genotypes.txt',
select = c(1),
header = F,
sep = "\t",
colClasses = c('character'))
names(genotype)[1] <- 'PigID'
ped <- vroom("Full_Pedigree.csv",
col_types = 'cccc')
pig_info <- vroom("PigInformation.csv",
col_types = 'cccccccc')
map <- vroom("50K_Porcine_n49991_header_20190528.map",
col_types = 'ccc')
### Remove pigs with IDs that begin in "G"
ped$filter <- grepl("G", ped$PigID)
ped <- ped %>%
filter(filter == F) %>%
select(PigID:BirthDate)
setwd("C:/Users/cgroh/OneDrive/Desktop/PhD/Project 1 - GSM/Intermediate Data/")
### Save as an .RData object
save(ped, pig_info, genotype, map, file = "RFiles.RData")Now, each time the code in this chapter is run, R will only need to load the “RFiles.RData” object, which is much quicker than reading in each file independently.
3.4 Data Dictionary
3.4.1 ped
The code used to present a glimpse, build the data dictionary, and describe each variable for ped is shown in the following three code chunks. In order to optimize the length of each chapter, code will only be shown if a certain procedure has not been presented before. However, the entire R script for each chapter will be available as separate files in an Appendix section.
## # A tibble: 6 x 4
## PigID SirePigID DamPigID BirthDate
## <chr> <chr> <chr> <chr>
## 1 13 98990 33776 201401
## 2 14 98990 33776 201401
## 3 15 98990 33776 201401
## 4 16 98990 33776 201401
## 5 17 98990 33776 201401
## 6 18 98990 33776 201401The ped dataset has 4 columns. Column descriptions are below:
### Describe variables
variable_names <- names(ped)
variable_description <- c("Progeny identification",
"Sire identification",
"Dam identification",
"Birth year and month (YYYYMM)")
variable_types <- c(class(ped$PigID),
class(ped$SirePigID),
class(ped$DamPigID),
class(ped$BirthDate))
### Build dictionary
dictionary <- tibble(`Variable Names` = variable_names,
Description = variable_description,
`Variable Types` = variable_types)
### Display dictionary
kable(dictionary,
caption = "Data dictionary for ped dataset.",
booktabs = TRUE,
row.names = FALSE)| Variable Names | Description | Variable Types |
|---|---|---|
| PigID | Progeny identification | character |
| SirePigID | Sire identification | character |
| DamPigID | Dam identification | character |
| BirthDate | Birth year and month (YYYYMM) | character |
The ped dataframe is the actual pedigree used in The Maschhoff’s routine genetic evaluation. For selection mapping analyses in the current project, only pigs in this pedigree matter. A summary of each variable is presented below:
## ped
##
## 4 Variables 1333508 Observations
## -------------------------------------------------------------------------------------------------------------------------------------------
## PigID
## n missing distinct
## 1333508 0 1333508
##
## lowest : 1 10 100 1000 10000 , highest: 999995 999996 999997 999998 999999
## -------------------------------------------------------------------------------------------------------------------------------------------
## SirePigID
## n missing distinct
## 1333508 0 4183
##
## lowest : 0 1002406 10035 10085 101789 , highest: 99925 99926 99927 99928 99929
## -------------------------------------------------------------------------------------------------------------------------------------------
## DamPigID
## n missing distinct
## 1333508 0 56522
##
## lowest : 0 1000199 1000233 1000327 1000329, highest: 999837 999839 999840 999841 999842
## -------------------------------------------------------------------------------------------------------------------------------------------
## BirthDate
## n missing distinct
## 1333508 0 418
##
## lowest : 190001 195101 198006 198101 198109, highest: 202005 202006 202007 202008 202009
## -------------------------------------------------------------------------------------------------------------------------------------------From this summary, The Maschhoff’s pedigree has the following attributes:
- 1,333,508 progeny from 4183 sires and 56,522 dams
- Lowest values for sire and dam columns is 0
- Pigs with 0 in the sire and dam columns are founder animals
- Birth dates range from the year 1900 to 2020
- Pigs with a birth date of 1900 or 1951 are likely founder animals
- There are no missing values
- Each pig has pedigree information (other than founder animals)
3.4.2 pig_info
First 6 rows of the pig_info dataset:
## # A tibble: 6 x 8
## PigID SirePigID DamPigID BirthLitterID Line Family Gender CurrentSiteCode
## <chr> <chr> <chr> <chr> <chr> <chr> <chr> <chr>
## 1 1208582 665056 1144503 170939 9006 9006 Not Specified Male MRNS
## 2 1208588 665056 1144503 170939 9006 9006 Not Specified Male MRNS
## 3 1357380 638657 1231035 194642 9006 9006 Not Specified Male MRNS
## 4 1444826 805912 1322914 208627 9006 9006 Not Specified Male MRNS
## 5 1480262 795950 1322720 217065 9006 9006 Not Specified Male MRNS
## 6 1480265 795950 1322720 217065 9006 9006 Not Specified Male MRNSThere are 8 columns in this dataset, and they are defined below:
| Variable Names | Description | Variable Types |
|---|---|---|
| PigID | Progeny identification | character |
| SirePigID | Sire identification | character |
| DamPigID | Dam identification | character |
| BirthLitterID | Unique identification of each litter in which the pig was born | character |
| Line | Genetic line, specified by The Maschhoff’s | character |
| Family | Family within genetic line, specified by The Maschhoff’s | character |
| Gender | Sex of the pig | character |
| CurrentSiteCode | Four digit site code for the current location of the pig within The Maschhoff’s | character |
The pig_info dataset expands upon progeny within the ped dataset by providing additional information about each pig. In subsequent sections, pigs that are in the pig_info dataset but not in the ped dataset will be filtered from pig_info. A summary of each column is displayed below:
## pig_info
##
## 8 Variables 1914836 Observations
## -------------------------------------------------------------------------------------------------------------------------------------------
## PigID
## n missing distinct
## 1914836 0 1914836
##
## lowest : 1 10 100 1000 10000 , highest: 999995 999996 999997 999998 999999
## -------------------------------------------------------------------------------------------------------------------------------------------
## SirePigID
## n missing distinct
## 1878403 36433 6060
##
## lowest : 1000683 1000684 1000685 1000686 1000687, highest: 99925 99926 99927 99928 99929
## -------------------------------------------------------------------------------------------------------------------------------------------
## DamPigID
## n missing distinct
## 1880853 33983 74584
##
## lowest : 1000199 1000200 1000201 1000205 1000206, highest: 999837 999839 999840 999841 999842
## -------------------------------------------------------------------------------------------------------------------------------------------
## BirthLitterID
## n missing distinct
## 1850771 64065 166060
##
## lowest : 1 1000 10002 10003 10004, highest: 9995 9996 9997 9998 9999
## -------------------------------------------------------------------------------------------------------------------------------------------
## Line
## n missing distinct
## 1914836 0 109
##
## lowest : 0 10 1005 1006 1007 , highest: Meishan 1 Meishan 2 P PL W
## -------------------------------------------------------------------------------------------------------------------------------------------
## Family
## n missing distinct
## 1914836 0 131
##
## lowest : 0 Not Specified 10 Not Specified 1005 Not Specified 1006 Not Specified 1007 Not Specified
## highest: Meishan 1 Not Specified Meishan 2 Not Specified P Not Specified PL Not Specified W Not Specified
## -------------------------------------------------------------------------------------------------------------------------------------------
## Gender
## n missing distinct
## 1914376 460 2
##
## Value Female Male
## Frequency 957768 956608
## Proportion 0.5 0.5
## -------------------------------------------------------------------------------------------------------------------------------------------
## CurrentSiteCode
## n missing distinct
## 1909484 5352 126
##
## lowest : 12345 4NL2 AMCO AVAS BC1S , highest: WLDV WTSR WVRD YDRM YDRN
## -------------------------------------------------------------------------------------------------------------------------------------------Key attributes of the pig_info dataset from the summary above are highlighted below:
- 1,914,836 progeny from 6060 sires and 74,584 dams (Reminder: only pigs in
pedmatter for this project) - 131 unique families within 109 unique genetic lines (Note: some lines do not have any family subsets; these are “Not Specified”)
- Proportions of males and females are equal (0.50)
- 126 unique locations
- Columns with missing data (Note: many of the rows that contain missing data will be filtered from this dataset, as they are unnecessary):
- SirePigID
- DamPigID
- BirthLitterID
- Gender
- CurrentSiteCode
3.4.3 genotype
Due to the size of the genotype file (> 1 GB), manipulation and analysis of this data is extremely difficult in R, especially on a local machine (laptop). Therefore, an example of the dataframe structure of genotype will be generated. This will be separate from the genotype dataframe, which is simply a vector of Pig ID’s that are genotyped.
## # A tibble: 5 x 2
## PigID Genotype
## <dbl> <chr>
## 1 1 0,2,0,1,5
## 2 2 1,1,0,5,2
## 3 3 2,1,0,5,1
## 4 4 2,2,2,5,1
## 5 5 1,1,2,1,0This example dataset (temp_genotype) has 5 pigs, each with 5 SNP positions. For consistency, a data dictionary of temp_genotype is presented below:
| Variable Names | Description | Variable Types |
|---|---|---|
| PigID | Pig identification | numeric |
| Genotype | Genotype string | character |
In this example dataset, the Genotype column is a string of haplotypes, with the haplotype for each SNP located between each comma. Since there are 49,991 SNPs in the real genotype dataframe, the Genotype column is 49,991 elements long. There are 4 possible haplotypes coded at each position:
- 0 = Alleles AA
- 1 = Alleles AB
- 2 = Alleles BB
- 5 = Missing genotype data
This SNP array is formatted for analysis in the BLUPF90 family of programs. Another common format is PLINK, which will be discussed in section 3.4.
In section 3.2, an outside R script was presented that read in the first column of the genotype file. For all subsequent analyses, this single column of Pig ID’s will be considered the genotype dataframe. A glimpse of genotype is presented below.
## # A tibble: 6 x 1
## PigID
## <chr>
## 1 0
## 2 12
## 3 166
## 4 196
## 5 204
## 6 206And a description of the single variable in the dataframe, PigID:
## genotype
##
## 1 Variables 66388 Observations
## -------------------------------------------------------------------------------------------------------------------------------------------
## PigID
## n missing distinct
## 66388 0 66388
##
## lowest : 0 1000199 1000201 1000226 1000233, highest: 999424 999431 999455 999494 999535
## -------------------------------------------------------------------------------------------------------------------------------------------A quick look at the output from the describe function reveals that 66,388 total pigs from the entire pedigree have been genotyped in The Maschhoff’s genetic evaluation program thus far. There are no missing values in this column, as expected.
3.4.4 map
One of the most important files in a genomic analysis is the MAP file, which connects each of the SNP columns in the genotype file to the actual genomic position in a certain reference genome. Coordinates in the MAP file used in this analysis are mapped to Sus scrofa 10.2. The first six rows of this file are shown below:
## # A tibble: 6 x 3
## CHR SNP_ID POS
## <chr> <chr> <chr>
## 1 1 ALGA0004882 100050454
## 2 1 DRGA0001340 100080552
## 3 1 MARC0070803 10010333
## 4 1 ALGA0004886 100142509
## 5 1 ALGA0004889 100165684
## 6 1 H3GA0002253 100223426And a description of each variable is presented below:
| Variable Names | Description | Variable Types |
|---|---|---|
| CHR | Chromosome number | character |
| SNP_ID | SNP name or ID number | character |
| POS | Physical base pair position | character |
These three columns provide all the necessary information to locate where each SNP in the genotype file sits across the pig genome. A summary of each variable is displayed below:
## map
##
## 3 Variables 49991 Observations
## -------------------------------------------------------------------------------------------------------------------------------------------
## CHR
## n missing distinct
## 49991 0 20
##
## lowest : 1 10 11 12 13, highest: 5 6 7 8 9
##
## Value 1 10 11 12 13 14 15 16 17 18 19 2 20 3 4 5 6 7 8 9
## Frequency 4917 1595 1836 1289 3643 3275 2865 1831 1408 1262 2323 3273 14 2897 3008 2335 3365 2825 2865 3165
## Proportion 0.098 0.032 0.037 0.026 0.073 0.066 0.057 0.037 0.028 0.025 0.046 0.065 0.000 0.058 0.060 0.047 0.067 0.057 0.057 0.063
## -------------------------------------------------------------------------------------------------------------------------------------------
## SNP_ID
## n missing distinct
## 49991 0 49991
##
## lowest : 1_10673082 1_10723065 1_11407894 1_11426075 1_13996200
## highest: WUR10000057 WUR10000071 WUR10000125 WUR10000125_2 WUR10000127
## -------------------------------------------------------------------------------------------------------------------------------------------
## POS
## n missing distinct
## 49991 0 49975
##
## lowest : 100004352 100012561 100014471 1000181 100019532, highest: 99980492 99985047 99989884 99992288 9999843
## -------------------------------------------------------------------------------------------------------------------------------------------The length of this dataset confirms that there are 49,991 SNPs in which genotypes are available for selection mapping analyses (before quality control, which is discussed later). A couple of other things to note:
- SNPs are not spread evenly across chromosomes. Chromosome 20 only has 14 SNPs, while chromosome 1 has 4917 SNPs
- There are no missing values
- The number of distinct POS values does not match the number of observations. These are not the only SNP identifiers (there are two other columns of information), so it likely will not affect the analysis
3.5 Data Preparation
Data preparation is a necessary task that takes on an infinitely many forms, depending on the goals of a project. Throughout this book, I am going to attempt to use a consistent system for getting data ready to be explored, mined, and modeled. However, some recurring terms will need to be defined before I begin.
3.5.1 Three Types of Data
- Raw Data: data that has never been manipulated; this data always needs to be stored in its original form, and the original file can NEVER be modified
- Tidy Raw Data raw data that has been modified to conform to Hadley Wickham’s definition of “tidy data”: 1) each row is an observation, 2) each column is a variable, and 3) each type of observational unit forms a table. Sometimes raw data and tidy raw data are the same, depending on the cleanliness of the raw data
- Intermediate Data: tidy raw data that has had anomalies and unnecessary observations and variables removed and additional variables calculated that are required in the analysis OR any new file or dataset that is created from tidy raw data; when data reaches the end of this stage, it is generally ready to be analyzed
3.5.2 Process Overview
Below is an outline of the data preparation process for each file:
ped
- Raw Data to Tidy Raw Data
- This file was communicated in tidy format
- Convert column types to correct class
- Tidy Raw Data to Intermediate Data
- Transfer BirthDate to
pig_info - Synch PigID, SirePigID, and DamPigID with
pig_info
- Transfer BirthDate to
pig_info
- Raw Data to Tidy Raw Data
- This file was communicated in tidy format
- Convert column types to correct class
- R object name changed to
ped_plus(pedplus pig information)
- Tidy Raw Data to Intermediate Data
- Remove pigs with a breed discrepancy (sire and dam breeds do not produce correct progeny breed combination)
- Remove unnecessary observations based on column Line
- Remove unnecessary observations based on CurrentSiteCode
- Create variables for birth date (from
ped), age, generation number, and whether or not a pig was genotyped (fromgenotype)
genotype
- Raw Data to Tidy Raw Data
- This file was communicated in BLUPF90 format, and the first column of pig ID’s were loaded into R in tidy format
- Convert column type to correct class
- Tidy Raw Data to Intermediate Data
- Create a column where each value is “TRUE” (for filtering purposes in
ped_plus)
- Create a column where each value is “TRUE” (for filtering purposes in
Note: data preparation using genotype will be included in the section for the pedigree files
map
- Raw Data to Tidy Raw Data
- This file was communicated in tidy format
- Convert column types to correct class
- Tidy Raw Data to Intermediate Data
- Reconcile “bad” entries in the POS column
genotypes.txt
Data preparation procedures were not limited to R in this section. Python and a combination of linux command line functions were used for handling the genotype files for each subgroup (given their size). PLINK was used to apply genotype quality control filters.
- Raw Data to Tidy Data
- Create subgroups based on subsequent analyses
- Convert genotype file for each subgroup to PLINK format
- Tidy Raw Data to Intermediate Data
- Perform genotype filtering for quality control on each subgroup
- Convert to BIM, BED, and FAM file formats for each subgroup
Additional Intermediate Data
- Genomic relationship matrices (GRMs) for each subgroup post-quality control using Genome-wide Complex Trait Analysis (GCTA)
- Phenotype files for each subgroup using R
3.5.3 Pedigree Files (ped and pig_info)
3.5.3.1 Filter by Genetic Line
The first step of the data preparation will be ensuring each variable in ped and pig_info are of the correct class. If we refer to the previous section, we can see the class of each column for these datasets in their respective data dictionaries is character. Whenever values are unknown in certain columns of raw data, it is best to read them in as type character, in order to preserve all values in the column. If we would attempt to read in a column as integers and some values in the column contain letters, those values will be removed. Nevertheless, all pig ID’s (progeny, sire, and dam) need to be integers, while all other columns need to be characters:
ped_tidy <- ped %>%
mutate_at(c(1:3), as.integer)
pig_info_tidy <- pig_info %>%
mutate_at(c(1:3), as.integer)The "_tidy" extensions was appended to ped and pig_info, which signifies they have passed all data quality checks required to be considered tidy data.
In the objectives of Chapter 2, it was stated that selection mapping analyses were going to be conducted for 4 genetic lines. The current ped and pig_info datasets, however, contain information on over 100 genetic lines. To get from the current state of the datasets to a point where they are ready for analysis, there are various data carpentry procedures that are required, such as row filtering, column selection, and variable calculation. There are 3 pure breeds that within these pedigree records, and they are as follows:
- Duroc (The Maschhoff’s Line 1006)
- Landrace (The Maschhoff’s Line 10 or 1005)
- Yorkshire (The Maschhoff’s Line 11)
Then, there are records on crossbred pigs:
- Crossbred [Duroc \(\times\) (Landrace \(\times\) Yorkshire)] (The Maschhoff’s Line 9006)
Why these 4 lines?
- Landrace, Yorkshire, and Duroc pigs are the backbone of the United States swine industry
- By far, pigs from these genetic backgrounds outnumber others in the genotype file
- Selection objectives are clear, defined by selection indices
Pedigree records in the pig_info_tidy dataset do not have breed assignments (only genetic lines). Below, breeds are assigned to each purebred pig.
### Merge to sync pedigree information and add birth date
ped_plus_tidy <- left_join(ped_tidy, pig_info_tidy, by="PigID") %>%
dplyr::mutate(SirePigID.x = SirePigID.y,
DamPigID.x = DamPigID.y,
PigBreed = ifelse(Line == 10 | Line == 1005, 'L',
ifelse(Line == 11, 'Y',
ifelse(Line == 1006, 'D', NA)))) %>%
rename(SirePigID = SirePigID.x,
DamPigID = DamPigID.x) %>%
select(PigID:BirthDate, PigBreed, Line:Gender, BirthLitterID, CurrentSiteCode)
### Deleted
rm(pig_info_tidy) The ped_tidy and ped_plus_tidy datasets contain 1,333,508 pigs. The name of pig_info_tidy was changed for clarity; because only pigs in the ped_tidy dataframe are going to be included in subsequent analyses, the pig_info_tidy dataframe was appended to ped_tidy, which made ped_plus_tidy (a dataframe of pedigree information plus pig information). Because there are over 1 million pigs (more chances for error), discrepancies between progeny genetic line and the genetic line combinations of the parents were checked and removed.
### Merge to create a column for breed of sire
ped_plus_tidy <- merge(ped_plus_tidy,
ped_plus_tidy[,c(1,5)],
by.x='SirePigID',
by.y = 'PigID',
all.x = TRUE) %>%
arrange(PigID) %>%
dplyr::rename(PigBreed = PigBreed.x,
SireBreed = PigBreed.y)
### Merge to create a column for breed of dam
ped_plus_tidy <- merge(ped_plus_tidy,
ped_plus_tidy[,c(2,5)],
by.x='DamPigID',
by.y = 'PigID',
all.x = TRUE) %>%
arrange(PigID) %>%
dplyr::rename(PigBreed = PigBreed.x,
DamBreed = PigBreed.y)
### Remove any rows where breed of sire and dam are not valid for breed of offspring
ped_plus_tidy <- ped_plus_tidy %>%
dplyr::mutate(filter1 = ifelse(is.na(PigBreed) == FALSE,
ifelse(PigBreed == SireBreed & PigBreed == DamBreed, FALSE, TRUE), FALSE)) %>%
filter(filter1 == FALSE | is.na(filter1) == TRUE) %>%
select(PigID, SirePigID, DamPigID, PigBreed, SireBreed, DamBreed,
Line, Family, BirthDate, Gender, BirthLitterID, CurrentSiteCode)Eighty-six pigs (0.006% of the entire pedigree) had breed discrepancies and were removed, leaving 1,333,422 pigs in each dataset. However, there are only genotype records for 4 genetic lines [Duroc, Landrace (The Maschhoff’s Line 10), Yorkshire, and Crossbred]. Thus, any pig that is not of these genetic backgrounds can be removed using the dplyr function of the tidyverse family of packages.
### Remove unnecessary breeds
ped_plus_tidy <- ped_plus_tidy %>%
filter(Line %in% c('10', '11', '1006', '9006'))After the genetic line filter was applied, 1,267,652 pigs remain in the ped_plus_tidy dataset, and below is the current structure of ped_plus_tidy:
## PigID SirePigID DamPigID PigBreed SireBreed DamBreed Line Family BirthDate Gender BirthLitterID CurrentSiteCode
## 1 1 98257 98258 Y Y Y 11 11 Not Specified 201005 Female <NA> AVAS
## 2 2 98989 63615 D D D 1006 1006 Not Specified 201401 Male 6856 SFLD
## 3 3 98989 63615 D D D 1006 1006 Not Specified 201401 Male 6856 RIVR
## 4 4 98989 63615 D D D 1006 1006 Not Specified 201401 Male 6856 AVAS
## 5 5 98989 63615 D D D 1006 1006 Not Specified 201401 Male 6856 RIVR
## 6 6 98989 63615 D D D 1006 1006 Not Specified 201401 Male 6856 AVAS3.5.3.2 Filter by Current Location
In early discussions of the project, The Maschhoff’s specified that pigs from a recent purchase of outside genetics should be removed from all analyses as selection in these animals was different than the majority of the pigs in the pedigree. Below, pigs with a CurrentSiteCode of “WHHS” or “WHSS” are removed from ped_plus_tidy. It is important that we remove these pigs before calculation of generation number in the next section, as these animals may have different pedigree depth than the rest of The Maschhoff’s population.
3.5.3.3 Additional Variables
In the previous section, all rows were filtered from ped_tidy and ped_plus_tidy that were not needed for the selection mapping analyses. However, there are a few additional columns that need to be created or calculated, namely generation and age. Each pig has a birth date, however it is in a unconventional format (YYYYMM) when it needs to be an actual calendar date (YYYY-MM-DD) for age calculation and time series plotting.
### Make a month and year column from BirthDate
ped_plus_tidy <- ped_plus_tidy %>% extract(BirthDate,
into = c("Year", "Month"), "(.{4})(.{2})",
remove = F) %>%
### Set birth day to the first of the month
dplyr::mutate(Day = '01') %>%
### Change BirthDate to variable type Date
dplyr::mutate(BirthDate = ymd(paste(Year,'-',Month,'-',Day, sep = ""))) %>%
select(PigID:Year, Gender:CurrentSiteCode) %>%
rename(Sex = Gender)Now, pedigree age can be calculated. Pedigree age (AGE) is defined as the difference between each pig’s birth month and January 2006. The selection of January 2006 as the reference date is arbitrary; however, in this population, the number of pedigree records began to increase steadily around this date (this will be discussed later).
ped_plus_tidy <- ped_plus_tidy %>%
dplyr::mutate(Age = interval(ymd(20060101), BirthDate) %/% months(1))The next variable to be calculated is generation number, which can be accomplished using the pedigree package. However, the pedigree needs to be sorted first.
### Order pedigree - parents before offspring
ord <- orderPed(ped_plus_tidy[,1:3])
ped_plus_tidy <- ped_plus_tidy[order(ord),]
### Calculate and assign generation number
gen <- countGen(ped_plus_tidy[,1:3])
ped_plus_tidy$Generation <- gen
### Append generation number to ped_plus_tidy dataframe, remove excess columns, and reorder
ped_plus_tidy <- left_join(ped_plus_tidy, ped_tidy[,1:3], by="PigID") %>%
dplyr::mutate(SirePigID.x = SirePigID.y,
DamPigID.x = DamPigID.y) %>%
rename(SirePigID = SirePigID.x,
DamPigID = DamPigID.x) %>%
select(PigID:Generation)
### Synchronize pedigree files
ped_tidy <- ped_plus_tidy[,1:3]
### Deleted
rm(gen, ord) The last variable that needs to be created is a column that specifies whether or not a pig was genotyped.
### Create a column of TRUEs for filtering
genotype_tidy <- genotype %>%
mutate(PigID = as.integer(PigID),
Genotyped = TRUE)
### Join ped_plus_tidy and genotype dataframes and reorder
ped_plus_intmd <- left_join(ped_plus_tidy, genotype_tidy, by="PigID") %>%
mutate(Genotyped = ifelse(is.na(Genotyped) == TRUE, FALSE, TRUE)) %>%
select(PigID:CurrentSiteCode, Genotyped, Age:Generation)
### Sync ped to ped_plus
ped_intmd <- ped_plus_intmd[,1:3]
### Glimpse of new variables
head(ped_plus_intmd[,c(1:3, 9:10, 14:16)]) ## PigID SirePigID DamPigID BirthDate Year Genotyped Age Generation
## 1 98259 0 0 2007-02-01 2007 FALSE 13 0
## 2 98260 0 0 2007-02-01 2007 FALSE 13 0
## 3 98257 98259 98260 2008-08-01 2008 FALSE 31 1
## 4 167298 0 0 2002-01-01 2002 FALSE -48 0
## 5 167301 0 0 2001-06-01 2001 FALSE -55 0
## 6 97664 167298 167301 2003-07-01 2003 FALSE -30 1The "_intmd" extension replaced "_tidy" in both ped_tidy and ped_plus_tidy, which signifies that these datasets have reached the intermediate stage and are ready for use in the selection mapping analysis, tables, and figures.
3.5.4 MAP File
MAP files in genomic analyses are files (usually single-space separated text files) that “map” each SNP in the genotype file or SNP array to its physical position in a pig’s genome. For this selection mapping analysis, Sus scrofa 10.2 is the reference genome in which each SNP is mapped. The MAP file used in the selection mapping analyses will need to be in a certain format. The first program the MAP file will be run through is PreGSF90 of the BLUPF90 family of programs. According to Aguilar and Misztal (2014; PreGSF90/PostGSF90 README), the SNP map file should have a header with the following column names, in order:
- SNP_ID - identification of the SNP (alphanumeric)
- CHR - chromosome number (numeric), starting from 1
- POS - position bp (numeric)
Thus, minor changes to the 50K_Porcine_n49991_header_20190528.map (provided by Dr. Crum on 9/4/20) are required.
### Add permanent row index to avoid row permutation
map <- map %>%
dplyr::mutate(id = row_number())
head(map)## # A tibble: 6 x 4
## CHR SNP_ID POS id
## <chr> <chr> <chr> <int>
## 1 1 ALGA0004882 100050454 1
## 2 1 DRGA0001340 100080552 2
## 3 1 MARC0070803 10010333 3
## 4 1 ALGA0004886 100142509 4
## 5 1 ALGA0004889 100165684 5
## 6 1 H3GA0002253 100223426 6First, we notice that the order of the columns is incorrect for PreGSF90. In addition, the POS column was read in as “character”, and it should be integer. This means that at least 1 value is not in numeric or integer form.
## Warning: Problem with `mutate()` input `POS`.
## i NAs introduced by coercion
## i Input `POS` is `as.integer(POS)`.The error messages tell us that by running as.integer on the POS column of map, NA values were introduced. This means that there are some values in the POS column that are non-numeric. These will now be “NA” in map_tidy, as R does not know how to render an integer from a value that contains letters or special characters.
### Create filter for rows with NA values in POS column
map_tidy <- map_tidy %>%
dplyr::mutate(filter1 = is.na(POS))
### Filter rows from above
error_rows <- map_tidy %>%
filter(filter1 == T) %>%
select(-POS, -filter1)
### Join with original MAP file
error_rows <- left_join(error_rows, map[,c(3,4)], by="id")
head(error_rows)## # A tibble: 6 x 4
## id SNP_ID CHR POS
## <int> <chr> <chr> <chr>
## 1 9384 MARC0023716 12 56619202-56619210
## 2 9437 MARC0030450 12 58737215-58737222
## 3 10658 ASGA0105842 13 172601926-172601928
## 4 12145 MARC0054009 13 32309148-32309150
## 5 17786 M1GA0024791 15 150861922-150861932
## 6 18951 MARC0072559 15 62238854-62238861The source of the error in the POS column is clear. Instead of single integers, genomic positions of these error SNPs are specified as ranges. The following code chunk will address these errors.
### Separate the first and last number of range into own column
error_rows <- error_rows %>%
separate(col = POS, into = c("POS1", "POS2"), sep = "-") %>%
select(-POS2) %>%
dplyr::mutate(POS1 = as.integer(POS1)*-1)
### Make changes to MAP file
map_intmd <- left_join(map_tidy[,1:4], error_rows[,c(1,4)], by="id") %>%
arrange(id) %>%
dplyr::mutate(POS = ifelse(is.na(POS) == T, POS1, POS)) %>%
select(-id, -POS1)
head(map_intmd)## # A tibble: 6 x 3
## SNP_ID CHR POS
## <chr> <chr> <dbl>
## 1 ALGA0004882 1 100050454
## 2 DRGA0001340 1 100080552
## 3 MARC0070803 1 10010333
## 4 ALGA0004886 1 100142509
## 5 ALGA0004889 1 100165684
## 6 H3GA0002253 1 100223426Above, the error in the POS column was fixed by changing the range of genomic positions to a single negative integer that corresponded to the first value in the range of genomic positions \(\times\) -1. This affected only 19 out of 49,991 SNP positions (0.04%). A space-separated text version of map_intmd MAP file will be used in all subsequent selection mapping analyses.
3.5.5 Genotype File
The next portion of this section will be centered around preparing the genotype files for the selection mapping analyses. From this point on, all data preparation and carpentry was performed on a server hosted at the Animal Science Research Center on The University of Missouri campus. Due to the size of the genotype files, increased computing power was required (at least more than what my local machine can handle). The name and operating system of the server is MUG06 and CentOS 6.7, respectively. As mentioned in 3.5.2, a multitude of programs will be used on MUG06. Scripts utilized in this process can be extremely long and rather detailed; thus, examples of each script will be shown in this section and the entire scripts will be shown in the Appendix.
3.5.5.1 Subgroups
Up to 3 different analyses will be run as a part of the greater selection mapping project:
- Univariate Variance Component Estimation
- Bivariate Variance Component Estimation
- Genome-wide Association Analyses
At this point, I am not going to discuss the nature of these analyses any further (that will be the subject of Chapter 4). Different subgroups of pigs will be required for different analyses (and some subgroups may have multiple types of analyses run on them). These subgroups are defined in the table below:
| Subgroup | Genetic lines | Code and File Name Abbreviation |
|---|---|---|
| 1 | Crossbred | c |
| 2 | Duroc | d |
| 3 | Landrace | l |
| 4 | Yorkshire | y |
| 5 | Crossbred, Duroc | cd |
| 6 | Crossbred, Landrace | cl |
| 7 | Crossbred, Yorkshire | cy |
| 8 | Duroc, Landrace | dl |
| 9 | Duroc, Yorkshire | dy |
| 10 | Landrace, Yorkshire | ly |
| 11 | Duroc, Landrace, Yorkshire | dly |
| 12 | Crossbred, Duroc, Landrace, Yorkshire | cdly |
After the genotype file was copied to MUG06 using FileZilla, a Python script was invoked to separate the genotype file into the subgroups in the above table. An example of that script is shown below for subgroup 1, and the entire script and log is documented in A.1:
# /data/cjgwx7/tml_gpsm/scripts/python/CreateSubgroups.py
import pandas as pd
### Specify paths to necessary files for import
path_genotypes = "../../raw_data/level1/genotypes.txt"
path_ped_plus_intmd = "../../intmd/pedigree_map/ped_plus_intmd.txt"
### Specify paths for file export
path_s1 = "../../raw_data/level2/c.txt"
### Specify column names for input files
colnames_genotypes = ['PigID', 'SNPs']
colnames_ped_plus_intmd = ['PigID', 'Line']
### Read in genotype file and replace commas between SNP haplotypes
genotypes = pd.read_csv(path_genotypes,
sep = "\t",
names = colnames_genotypes,
header = None)
genotypes = genotypes.replace(',',
'',
regex=True)
### Read in ped_plus_intmd
ped_plus_intmd = pd.read_csv(path_ped_plus_intmd,
sep = " ",
names = colnames_ped_plus_intmd,
header = None,
usecols = [0, 6])
print("Data Imported Successfully")
### Merge genotype and ped_plus_intmd to connect PigID to Line information
genotypes_plus = pd.merge(genotypes,
ped_plus_intmd,
how = 'left',
on = 'PigID')
genotypes_plus = genotypes_plus[['PigID', 'Line']]
genotypes_plus = genotypes_plus.dropna()
### Create subgroups
## Crossbred
c = genotypes_plus[genotypes_plus.Line == 9006]
## Crossbred, Duroc, Landrace, and Yorkshire
cdly = genotypes_plus
cdly['f'] = cdly.Line.isin([9006, 1006, 10, 11])
cdly = cdly[cdly.f == True]
cdly = cdly.drop(columns=['f'])
print("Subgroups Created Successfully")
### Merge subgroup IDs with genotypes
c = pd.merge(c['PigID'], genotypes, how='left', on='PigID')
cdly = pd.merge(cdly['PigID'], genotypes, how='left', on='PigID')
print("Genotype File Merged Successfully")
### Check file length (Number of pigs before quality control)
print("The length of Subset 1 is:", len(c))
print("The length of Subgroup 12 is:", len(cdly))
print("Starting output of subgroup files")
c.to_csv(path_s1, sep = "\t", index = False, header = False)
cdly.to_csv(path_s12, sep = "\t", index = False, header = False)
print("Genotype files for each subgroup successfully written")3.5.5.2 BLUPF90 to PLINK
RENUMF90 will be the next program that each subgroup file will be run through in order to prepare them for PREGSF90, which will save the files in PLINK format (MAP and PED). Before I go any further, I need to explain PLINK PED and MAP files and their format [mainly how it is different than BLUPF90 (3.4.3)]. A PED file contains information about each subject (in our case, each pig) and all of their genotypes. Columns in this file are as shown below:
- Family ID (generally the same as individual ID in animals)
- Individual ID
- Paternal ID
- Maternal ID
- Sex (1 = male; 2 = female; other = unknown)
- Phenotype (quantitative; -9 = missing)
- Allele 1 for SNP 1
- Allele 2 for SNP 1
- Columns 7 and 8 are repeated for each SNP
The below table presents differences between PLINK PED and BLUPF90 genotypes:
| BLUPF90 | PLINK | Haplotype |
|---|---|---|
| 0 | 1 1 | AA |
| 1 | 1 2 | AB |
| 2 | 2 2 | BB |
| 5 | 0 0 | missing |
And lastly a screenshot of an example PED file:

Figure 3.1: Example of a PLINK PED file.
A PLINK MAP file is similar to a BLUPF90 MAP file, with a couple minor differences. Columns in a PLINK MAP file are described below:
- Chromosome number
- SNP ID
- Genetic distance (usually 0 in our case)
- Base-pair position
The only major differences between PLINK MAP and BLUPF90 MAP files are the addition of the genetic distance column and the order of the columns. However, the genetic distance column is usually always set to zero in our analyses, as we do not need the column.
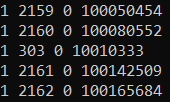
Figure 3.2: Example of a PLINK MAP file.
Now that we have the differences between PLINK and BLUPF90 genotype and map files sorted, we can get back to the preparation of the genotype data. There is a limit on the number of genotyped animals that can be input into a BLUPF90 program. Thus, each of the subgroup files that are greater than 25,000 will have to be split into smaller batches (everything but subgroups 1 through 4). The following bash script, B.1, will accomplish this and all subgroup and batch files will be output as fixed-width for BLUPF90 to properly read them:
# /data/cjgwx7/tml_gpsm/scripts/bash/batch.sh
### Create fixed-width BLUPF90 genotype file for each subgroup
awk -F ' ' '{printf "%-15s%-50000s\n",$1,$2}' ../../raw_data/level2/c.txt > ../../raw_data/level2/c.txt_fw
awk -F ' ' '{printf "%-15s%-50000s\n",$1,$2}' ../../raw_data/level2/cdly.txt > ../../raw_data/level2/cdly.txt_fw
### Delete old .txt files
rm ../../raw_data/level2/*.txt
### Split files into batches of < 25,000 observations each
### Subgroup 1
cp ../../raw_data/level2/c.txt_fw ../../raw_data/level3/c/c.aa
### Subgroup 12
split --verbose ../../raw_data/level2/cdly.txt_fw -l21015 cdly.
mv cdly* ../../raw_data/level3/cdly/Now, all batch files within each subgroup are in a fixed-width column format and have less than 25,000 observations each. Thus, we are now ready to build the parameter files for RENUMF90. Each batch file within each subgroup will be run through RENUM90 separately. An example of a RENUMF90 parameter file (essentially the file that is input into the program), for batch “aa” in subgroup 1 (Crossbred pigs) is presented below:
DATAFILE
ped_plus_intmd.txt
TRAITS
14
FIELDS_PASSED TO OUTPUT
1
WEIGHT(S)
RESIDUAL_VARIANCE
2.0
EFFECT # 1st effect: animal
1 cross alpha
RANDOM ## treated as a random effect
animal
FILE
ped_intmd.txt
FILE_POS
1 2 3 0 0
SNP_FILE ## SNP marker file
c.aa
(CO)VARIANCES ## its variance component
0.5
OPTION map_file map_intmd.txt
OPTION savePLINK
OPTION no_quality_controlThe parameter files for each batch will be incorporated in the following bash script (B.2; only an example is shown below), named “renum.sh”:
# /data/cjgwx7/tml_gpsm/scripts/bash/renum.sh
### Subgroup 1
### Copy necessary files into current directory
cp ../../intmd/pedigree_map/{ped_plus_intmd.txt,ped_intmd.txt,map_intmd.txt} .
### Copy genotype file into current directory
cp ../../raw_data/level3/c/c.aa .
### Run renumF90
renumf90 ../blupf90/renum.c_aa
### Save PLINK PED and MAP files
preGSf90 renf90.par
### Move important files to new location
mv renf90.dat toPlink.map toPlink.ped renadd01.ped c.aa_XrefID sum2pq freqdata.count Gen_call_rate ../../raw_data/level4/c/
### Remove unnecessary files
rm c.aa renf90.tables renf90.fields renf90.par
### Repeat above for each subgroup/batch combination
### Subgroup 12
cp ../../raw_data/level3/cdly/cdly.aa .
renumf90 ../blupf90/renum.cdly_aa
preGSf90 renf90.par
mv renf90.dat toPlink.map toPlink.ped renadd01.ped cdly.aa_XrefID sum2pq freqdata.count Gen_call_rate ../../raw_data/level4/cdly/aa/
rm cdly.aa renf90.tables renf90.fields renf90.par
cp ../../raw_data/level3/cdly/cdly.ab .
renumf90 ../blupf90/renum.cdly_ab
preGSf90 renf90.par
mv renf90.dat toPlink.map toPlink.ped renadd01.ped cdly.ab_XrefID sum2pq freqdata.count Gen_call_rate ../../raw_data/level4/cdly/ab/
rm cdly.ab renf90.tables renf90.fields renf90.par
cp ../../raw_data/level3/cdly/cdly.ac .
renumf90 ../blupf90/renum.cdly_ac
preGSf90 renf90.par
mv renf90.dat toPlink.map toPlink.ped renadd01.ped cdly.ac_XrefID sum2pq freqdata.count Gen_call_rate ../../raw_data/level4/cdly/ac/
rm cdly.ac renf90.tables renf90.fields renf90.par
### Remove unnecessary files
rm ped_plus_intmd.txt ped_intmd.txt map_intmd.txtThe script above is accomplishing numerous tasks, listed below:
- Copy ped_plus_intmd.txt, ped_intmd.txt, and map_intmd.txt files into the current directory (where the renum.sh script is stored and will be run)
- Copy genotype file into the current directory
- Run RENUMF90
- Run PREGSF90, using the renf90.par file generated by RENUMF90
- Move necessary output from PREGSF90 (PLINK files and renumbered pedigree and data files) to the correct subgroup and batch directory in the raw_data folder
- Remove unnecessary output files
- Remove files copied in step 1 from the script directory
The main file of interest generated from renum.sh is the toPlink.ped file for each batch within subgroup. This is a PED file, as described above. However, there are three issues with these files:
- Animal, sire, and dam IDs were renumbered through RENUMF90
- Sex is incomplete
- An extra blank column was added between the first 6 columns and the genotype data
The following script (B.3) will fix these errors, along with row-binding each batch file into a single PED file for each subgroup:
# /data/cjgwx7/tml_gpsm/scripts/bash/ProcessPlinkF90.sh
### Subgroup 1
### Remove extra blank column
cut -d ' ' -f 1-6,8- ../../raw_data/level4/c/toPlink.ped > ../../raw_data/level4/c/toPlink_v2.ped
### Extract genotypes
cut -d ' ' -f 7- ../../raw_data/level4/c/toPlink_v2.ped > ../../raw_data/level4/c/toPlink_geno.ped
### Replace renumbered IDs with actual IDs
Rscript ../r/ProcessPlinkF90/c_aa.R
### Column bind columns 1-6 and the genotypes
paste -d ' ' ../../raw_data/level4/c/c_aa.fam ../../raw_data/level4/c/toPlink_geno.ped > ../../raw_data/level6/c.ped
### Subgroup 12
### Batch 1
cut -d ' ' -f 1-6,8- ../../raw_data/level4/cdly/aa/toPlink.ped > ../../raw_data/level4/cdly/aa/toPlink_v2.ped
cut -d ' ' -f 7- ../../raw_data/level4/cdly/aa/toPlink_v2.ped > ../../raw_data/level4/cdly/aa/toPlink_geno.ped
Rscript ../r/ProcessPlinkF90/cdly_aa.R
paste -d ' ' ../../raw_data/level4/cdly/aa/cdly_aa.fam ../../raw_data/level4/cdly/aa/toPlink_geno.ped > ../../raw_data/level5/cdly/cdly_aa.ped
### Batch 2
cut -d ' ' -f 1-6,8- ../../raw_data/level4/cdly/ab/toPlink.ped > ../../raw_data/level4/cdly/ab/toPlink_v2.ped
cut -d ' ' -f 7- ../../raw_data/level4/cdly/ab/toPlink_v2.ped > ../../raw_data/level4/cdly/ab/toPlink_geno.ped
Rscript ../r/ProcessPlinkF90/cdly_ab.R
paste -d ' ' ../../raw_data/level4/cdly/ab/cdly_ab.fam ../../raw_data/level4/cdly/ab/toPlink_geno.ped > ../../raw_data/level5/cdly/cdly_ab.ped
### Batch 3
cut -d ' ' -f 1-6,8- ../../raw_data/level4/cdly/ac/toPlink.ped > ../../raw_data/level4/cdly/ac/toPlink_v2.ped
cut -d ' ' -f 7- ../../raw_data/level4/cdly/ac/toPlink_v2.ped > ../../raw_data/level4/cdly/ac/toPlink_geno.ped
Rscript ../r/ProcessPlinkF90/cdly_ac.R
paste -d ' ' ../../raw_data/level4/cdly/ac/cdly_ac.fam ../../raw_data/level4/cdly/ac/toPlink_geno.ped > ../../raw_data/level5/cdly/cdly_ac.ped
### Row bind each batch file
cat ../../raw_data/level5/cdly/cdly_aa.ped ../../raw_data/level5/cdly/cdly_ab.ped ../../raw_data/level5/cdly/cdly_ac.ped > ../../raw_data/level6/cdly.pedWithin the above script, R scripts (Ex: c_aa.R) are utilized to convert renumbered IDs (from BLUPF90) back to the actual pig IDs. An example of this script, for subgroup 1, is shown below:
library(data.table)
library(dplyr)
rm(list=ls())
path_ped_plus_intmd <- '../../intmd/pedigree_map/ped_plus_intmd.csv'
path_fam <- '../../raw_data/level4/c/toPlink.ped' # CHANGE
path_renped <- '../../raw_data/level4/c/renadd01.ped' # CHANGE
path_out <- '../../raw_data/level4/c/c_aa.fam' # CHANGE
ped_plus_intmd <- fread(path_ped_plus_intmd,
header = T,
sep = ",",
select = c("PigID", "Sex"))
fam <- fread(path_fam,
header = F,
sep = " ",
col.names = c('FamID', 'PigID', 'SirePigID', 'DamPigID', 'Sex', 'Phenotype'),
select = c(1:6)) %>%
tibble::rowid_to_column("index")
renped <- fread(path_renped,
header=F,
sep = " ",
select = c(1,10),
col.names = c('RenID', 'PigID'))
fam_v2 <- merge(fam, renped,
by.x = "PigID", by.y = "RenID",
all.x = TRUE) %>%
select(-PigID) %>%
dplyr::rename(PigID = PigID.y)
fam_v3 <- merge(fam_v2, renped,
by.x = "SirePigID", by.y = "RenID",
all.x = TRUE) %>%
select(-SirePigID) %>%
dplyr::rename(PigID = PigID.x,
SirePigID = PigID.y)
fam_v4 <- merge(fam_v3, renped,
by.x = "DamPigID", by.y = "RenID",
all.x = TRUE) %>%
select(-DamPigID) %>%
dplyr::rename(PigID = PigID.x,
DamPigID = PigID.y)
fam_v4 <- fam_v4[,c(2, 5:7, 3:4, 1)]
fam_v5 <- left_join(fam_v4, ped_plus_intmd, by="PigID") %>%
mutate(Sex.x = ifelse(Sex.y == "Male", 1, ifelse(Sex.y == "Female", 2, 0)),
FamID = PigID,
SirePigID = ifelse(is.na(SirePigID) == T, 0, SirePigID),
DamPigID = ifelse(is.na(DamPigID) == T, 0, DamPigID),
Phenotype = -9) %>%
arrange(index) %>%
select(-c(Sex.y, index)) %>%
dplyr::rename(Sex = Sex.x)
fwrite(fam_v5,
path_out,
row.names = FALSE,
col.names = FALSE,
sep = " ",
quote = FALSE)Again, a lot of different processes are happening in the above two scripts. The following procedure is completed for i = aa, ab, or ac (depending on how many batches are in a subgroup) and j = c, d, l, y, cd, cl, cy, dl, dy, ly, dly, or cdly:
- Column 7 (blank column) is removed from batch file i from subgroup file j
- Batch file i of subgroup file j is split into two files (toPlink_v2.ped: Columns 1-6; toPlink_geno.ped: Columns 7 to end)
- Note: File 1 is in FAM format of PLINK
- R script for batch file i of subgroup j matches renumbered IDs with original IDs from the ped_plus_intmd.txt file and outputs the FAM file for each i of j
- The FAM file is “column-binded” with the toPlink_geno.ped for batch file i from subgroup file j to create file j_i.ped
- Within subset j, files j_i.ped are row-binded into one file and stored in “/data/cjgwx7/tml_gpsm/raw_data/level6/” as j.ped (the final PED file for each subgroup)
3.5.5.3 PED Binary Conversion
The resulting files from the above procedure are now ready for input into PLINK to create BED, BIM, and FAM files and for quality control of at the variant (SNP) and pig level. A FAM file is simply the first 6 columns of a PED file, a BIM file is the MAP file for each subgroup (after quality control), and a BED file is the genotype part of a PED file in binary format, which uses less memory and is quicker for the computer to read. In addition, we only want to use autosomal SNPs, which are only on chromosomes 1 to 18. The below script (B.4) performs this conversion and removes the X and Y chromosomes (for subgroup 1):
3.5.5.4 Quality Control
The last modification of the genotype files will be filtering for genotype quality control. The following filters will be applied using PLINK:
- Remove SNPs with a genotype call rate less than 90%
- Remove SNPs with a minor allele frequency (MAF) less than 0.01
- Remove individual pigs with genotype call rates less than 90%
The bash script below (B.5) performs genotype filtering for quality control in each subgroup. The BED, BIM, and FAM files that are output from this script will be intermediate files used in all subsequent analyses.
# /data/cjgwx7/tml_gpsm/scripts/bash/
### Filter by SNPs
### Subgroup 1
plink --bfile ../../raw_data/level7/c/c --out ../../intmd/input_data/c/c --make-bed --maf 0.01 --geno 0.1 --nonfounders
### Filter by pigs
plink --bfile ../../intmd/input_data/c/c --out ../../intmd/input_data/c/c --make-bed --mind 0.1 --nonfoundersWith the conclusion of genotype quality control filtering, the genotype files for each subgroup are now ready for genomic analyses. However, two more files for each subgroup are required, and these will be discussed in the next section.
3.5.6 Additional Files
3.5.6.1 Genomic Relationship Matrices
A genomic relationship matrix (GRM) is a matrix that estimates the genomic similarity between each animal in a genotype file based on their SNP haplotypes. GCTA has a function to estimate this matrix based off of the method described by Yang et al., 2010. Below is the command that was invoked to produce a GRM for subgroup 1 (entire bash script is in B.6):
3.5.6.2 Phenotype Files
The last file required for the genomic analyses is a phenotype file for each subgroup. Each subgroup will need at least one phenotype file with the following columns:
- Family ID
- Individual ID
- Phenotype (in this case, AGE)
Subgroups that will be used in a bivariate analysis, however, will need an additional phenotype file with the following columns:
- Family ID
- Individual ID
- Phenotype Trait 1 (Age for the first population)
- Phenotype Trait 2 (Age for the second population)
Below is an example of this script (ran in R; C.1) for subgroup 5 (Crossbred and Duroc populations), which will show how each phenotype filetype is made:
library(data.table)
library(dplyr)
rm(list=ls())
### Global files
path_ped_plus_intmd <- '../../intmd/pedigree_map/ped_plus_intmd.txt'
ped_plus_intmd <- fread(file = path_ped_plus_intmd,
sep = " ",
header = F,
select = c(1, 7, 15),
col.names = c('PigID', 'Line', 'Age'))
### Subgroup 1
path_fam_c <- '../../intmd/input_data/c/c.fam'
path_c_uni_out <- '../../intmd/phenotype/c/c_uni.pheno'
fam_c <- fread(file = path_fam_c,
sep = " ",
header = F,
select = c(1:2),
col.names = c("FID", "PigID")) %>%
tibble::rowid_to_column()
pheno_c_uni <- left_join(fam_c, ped_plus_intmd[,c(1, 3)], by="PigID") %>%
arrange(rowid) %>%
select(-rowid)
fwrite(pheno_c_uni,
file = path_c_uni_out,
sep = " ",
row.names = FALSE,
col.names = FALSE,
quote = FALSE,
na = "NA") Finally, with all phenotype files made, we have everything we need to conduct genomic analyses. Thus, th data preparation is complete. As the analyses proceed, additional files or modifications to the intermediate files may be required; however, Chapter 3 provides an extremely detailed overview of how to turn raw genomic data into tidy files that are ready for analyses. The next chapter, Chatper 4, will describe the nuts and bolts of each genomic analysis.