7 Finding Genes by Sequence Variant
A gene is a segmant of DNA that is first transcribed to produce a gene product. In chapters 3 and 4, we looked at RNA to pinpoint the location of a gene. A gene is also “a functional unit of inheritance”. In this chapter you will see how DNA variants57 can be used to locate genes. How? Some DNA variants in humans are pathogenic (cause disease). These variants are assumed to inactivate a gene somehow underscoring the importance of that nucleotide or nucleotides within the gene. When the DNA variant is mapped to the genome, the location of the disrupted gene is also discovered. To follow along with the text and answer Test Your Understanding questions, you will be using the “DNA Variant” session link. You can also use this link as a starting point to view DNA variant data for any gene of interest.
7.1 Understanding how DNA variants are displayed
Open the “DNA Variant” session link to view the “ClinVar Variants” evidence track. This evidence track is configured to display two subtracks entitled, “ClinVar Short Variants < 50bp” (top) and “ClinVar SNVs submitted interpretations and evidence” (bottom). Both subtracks show the genomic positions of DNA substitutions58 and indels59 from the ClinVar database. “ClinVar is a free, public archive of reports of the relationships among human variations and phenotypes, with supporting evidence.”60. Importantly, the DNA variants included in each subtrack come from a similar but nonoverlapping set of data.
The DNA variants in both subtracks are color coded according to clinical significance: red for pathogenic (P) or likely pathogenic (LP), green for benign (B) or likely benign (LB), dark blue for “variant of uncertain significance” (VUS), dark gray for not provided (OTH) and light blue for “conflicting interpretation of pathogenicity”. In the “ClinVar Short Variants < 50bp” subtrack, each DNA variant is also displayed according to variant type. See Figure 7.1 for a list of DNA variant types you are likely to encounter.
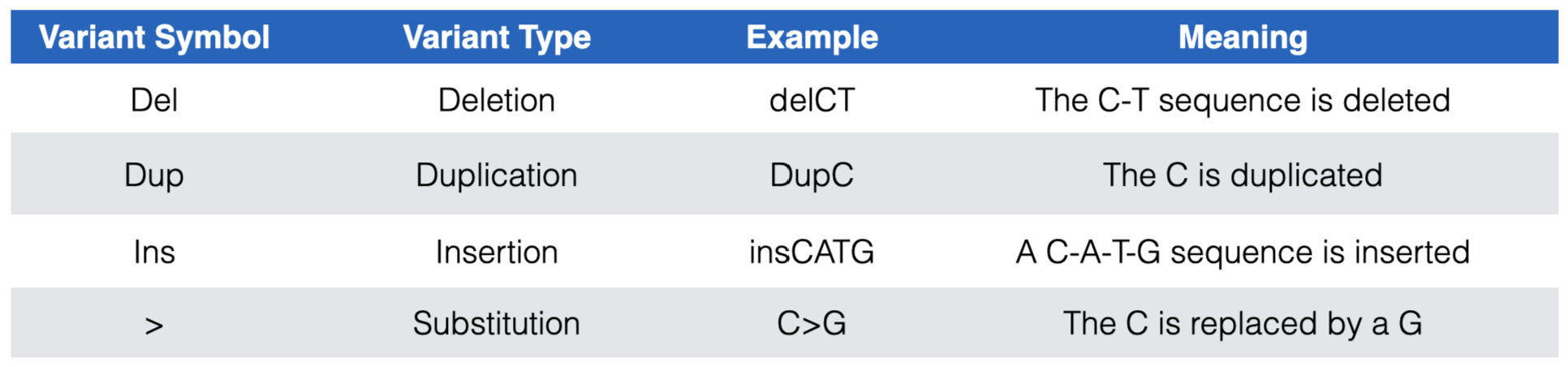
Figure 7.1: The most common DNA variant symbols you are likely to see in the ClinVar Short Variants < 50 bp substrack.
Each DNA variant in the “ClinVar Short Variants < 50bp” subtrack is clickable. A mouse click will take you to a new page with a table that describes the sequence variant in more detail. Useful information can often be found in the rows entitled, “Link to ClinVar with Variant ID”, “Clinical significance”, “Molecular Consequence” and “Phenotypes”. The “Link to ClinVar with Variant ID” row tells you exactly which nucleotide in the gene is affected and how this DNA change impacts the mRNA sequence and the protein sequence (if at all or if known). Figure 7.2 lists common DNA variant IDs you may encounter. These examples all map to CTSF, a gene nearby BBS1 (zoom out to locate it). For example, you might find “NM_003793.4(CTSF):c.649C>T (p.Arg217Ter)”. This means that that the C nucleotide, in the reference genome61 at position 649 of the CTSF mRNA (mRNA sequence described in NM_003793.462) is changed to a T. As a result of this DNA substitution, the reference amino acid at position 217 of the protein changes from an Arg (Arginine) to a termination codon (stop codon). Click here to review how DNA variant types are classified at both the DNA and protein levels.
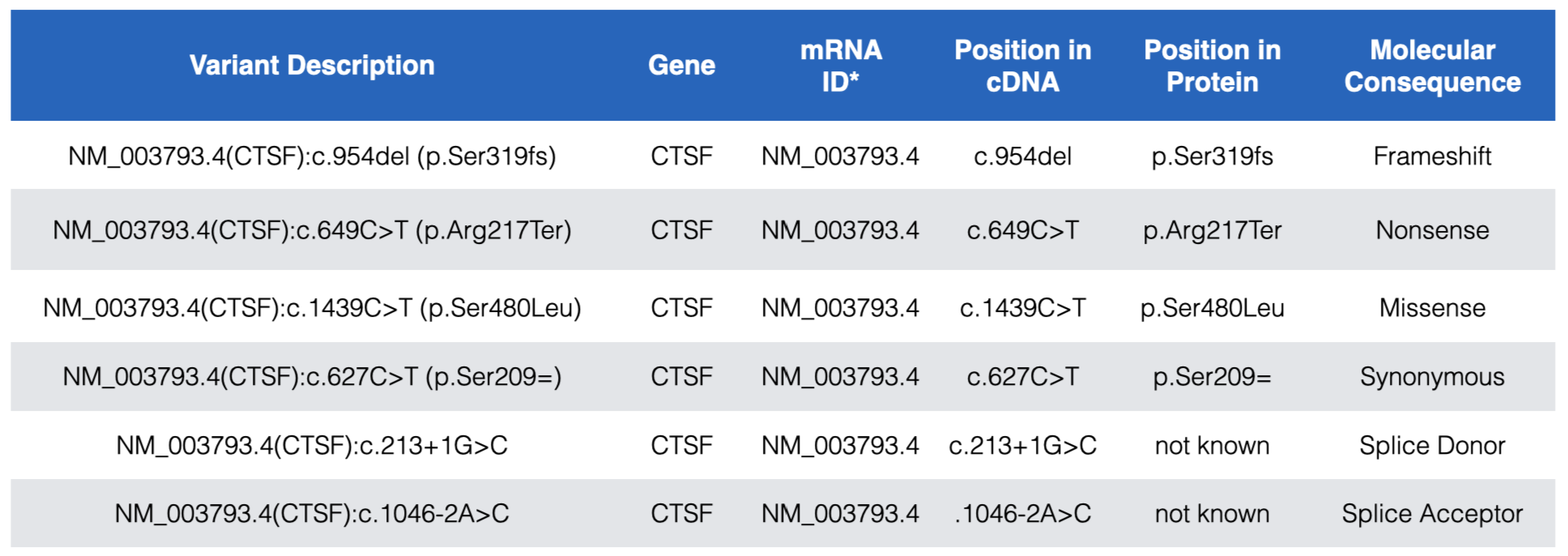
Figure 7.2: Examples of the most common DNA variants and how the Variant description explains where each maps within the gene, mRNA (cDNA) and protein. In the Variant Description and Position in Protein columns, Ter=termination and the equal sign indicates no change. Also notice that while splice site variants do not map to coding sequence they are still described relative to a position in the cDNA. See text for more details.
You can also click on any circle (red, green, etc) in the ClinVar Interpretations subtrack to review a similar table of information except that the “Phenotype” row is replaced by a “Description” row. This row typically explains the impact that the DNA variant has on the gene product and the person harboring the variant63. Although sometimes it is left blank.
As you explore BBS1 DNA variants, you will see that most pathogenic DNA variants map to coding sequence. One example is shown in Figure 7.3. The reference codon, C-G-A, is outlined in red (See Base Position Track). C-G-A codes for Arginine (Arg or R) at position 146 (See Gene Prediction Track). The pathogenic mutation at this position is a C>T substitution changing the C-G-A codon to T-G-A. If you consult the genetic code (Figure 2.4) you will see that T-G-A codes for a stop codon. This is consistent with the classification of this variant as pathogenic.
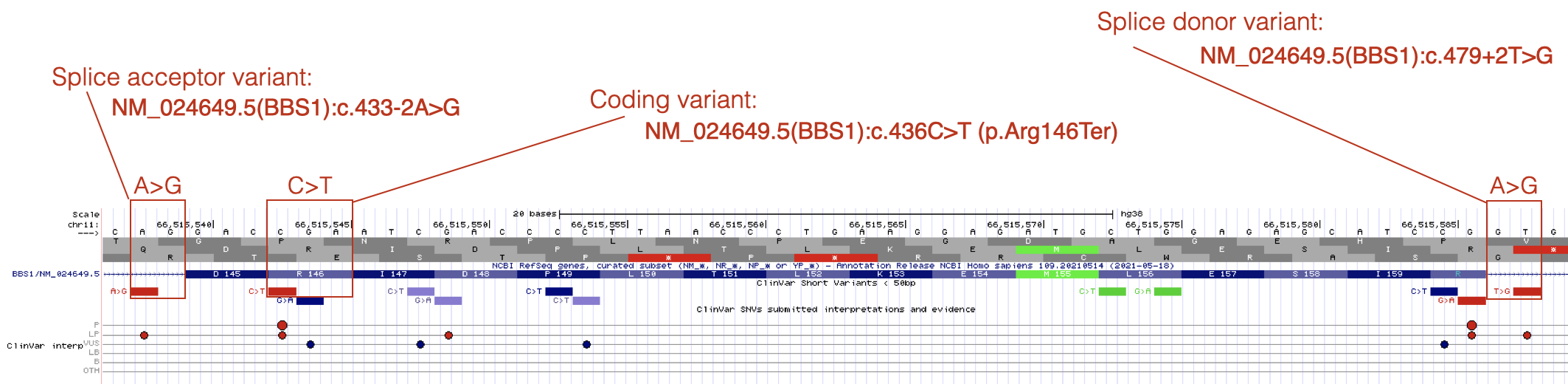
Figure 7.3: Three variants that are described in detail in the text.
By contrast, pathogenic mutations that map outside the coding sequence typically map to either a 5’ splice site (splice donor site) or a 3’ splice site (splice acceptor site). An example of each is also shown in Figure 7.3. Notice that DNA variants that map to splice sites do not provide information about how the DNA variant impacts the protein sequence (click on any splice site mutation and review the Clinvar Variant ID or Protein HGVS64 information). This is because it is nearly impossible to predict how a splice site mutation will impact splicing and thus the main open reading frame (main ORF) that is produced. The intron might remain in the mature mRNA, a nearby cryptic splice site65 might get used instead or the exon impacted by the splice site mutation might be skipped entirely. Whatever happens, splice site mutations typically lead to shifts in the reading frame and early stop codons.
Finally, notice how the position of a splice site mutation is described relative to the mRNA (cDNA). The location of the splice acceptor variant in Figure 7.3 is described as NM_024649.5(BBS1):c.433-2A>G. In this example, position 433 is the first nucleotide of exon 5 of BBS1 (as described by NM_024649.5). Thus the DNA variant is located at position 433-2 (the 2nd nucleotide upstream of exon 5 in the genome). In other words, the highly conserved A in the A-G of the 3’ SS of intron 4 is mutated to a G. The location of the splice donor variant in Figure 7.3 is described as NM_024649.5(BBS1):c.479+2T>G. In this example, position 479 is the last nucleotide of exon 5. Thus, the DNA variant is located at position 479+2 (the 2nd nucleotide downstream of exon 5). In other words, the highly conserved T in the G-T of the 5’ SS of intron 5 is mutated to a G.
Here is a useful trick. Given a Clinvar variant ID (i.e. NM_024649.5(BBS1):c.433-2A>G) or Nucleotide HGVS ID (i.e. NM_024649.4:c.433-2A>G) you can quickly and easily locate the DNA variant within the UCSC genome browser. Simply copy and paste the either ID type into the search window and click “Go”. This knowledge will come in handy when you complete your “Test Your Understanding” questions!
7.1.1 Test Your Understanding
To answer the following “Test Your Understanding” questions, use the “DNA Variant” session. You may also need to consult Figure 7.4 below to remember how amino acids are grouped according to the functional properties associated with their side chains (hydrophobic, polar uncharged etc).
Consider the coding variant NM_024649.5:c.235G>T to answer the following questions:
- Copy and paste Nucleotide HGVS description from above into the Genome Browser window. What type of sequence variant is this (i.e. transition, transversion or indel)?
- What is the sequence of the reference codon impacted by the G>T mutation?
- What amino acid does the reference codon code for?
- What does the reference codon become in the NM_024649.5:c.235G>T variant?
- What amino acid does the variant codon code for (if any)?
- What type of protein variant is this?
Consider the coding variant NM_024649.5:c.41C>G to answer the following questions:
- Copy and paste Nucleotide HGVS description from above into the Genome Browser window. What type of sequence variant is this (i.e. transition, transversion or indel)?
- What is the sequence of the reference codon impacted by the C>T mutation?
- What amino acid does the reference codon code for?
- What does the reference codon become with the NM_024649.5:c.41C>G variant?
- What amino acid does the variant codon code for (if any)?
- What type of protein variant is this?
Consider the coding variant NM_024649.5:c.240C>T to answer the following questions:
- Copy and paste Nucleotide HGVS description from above into the Genome Browser window. What type of sequence variant is this (i.e. transition, transversion or indel)?
- What is the sequence of the reference codon impacted by the C>T mutation?
- What amino acid does the reference codon code for?
- What does the reference codon become with the NM_024649.5:c.240C>T variant?
- What amino acid does the variant codon code for (if any)?
- What type of protein variant is this?
General Questions about mutations.
- What type of sequence variants lead to a frameshift mutation (transition, transversion, indel)?
- By definition, where must frameshift mutations be located (i.e. exons, introns, 5’ UTR, 3’ UTR, promoter)?
- By definition, where must splice site mutations be located (i.e. exons, introns, 5’ UTR, 3’ UTR, promoter)?
- By definition, where must nonsense mutations be located (i.e. exons, introns, 5’ UTR, 3’ UTR, promoter)?
Step back and look at a larger region surrounding BBS1. Specifically, click on the link for “DNA Variant” session then zoom out 10X. This region now includes neighboring genes including MRPL11, DPP3, BBS1, ZDHHC24, ACTN3 and CTSF.
- Copy and paste a nucleotide HGVS code for any frameshift mutation that maps to BBS1
- Pathogenic sequence variants map to two genes in this region. Choose the two among MRPL11, DPP3, BBS1, ZDHHC24, ACTN3 and CTSF?
.](static/images/aminoacidgroups.png)
Figure 7.4: Amino acid side chains can be grouped according to chemical properties. This image illustrates one way amino acids can be grouped. Image credit: Dan Cojocari.
7.2 For Discussion
- Some genes don’t have any variants associated with them. Why do you think this is? More than one answer is possible.
- Find the sequence variant that maps to the gene, CCS (Nucleotide HGVS: NM_005125.2:c.487C>T). It suggests that it might lead to neurodegeneration but it is rated as “uncertain significance”. How likely do you think it is that this is a pathogenic allele. Use any tool (including Google searches, your knowledge of genetics and reasoning abilities) to argue one way or the other (there are no right answers).
7.3 Caveats and Limitations
Absence of evidence is not evidence of absence. In other words, every gene cannot be identified in this way even if a DNA variant renders a gene inactive. Why, you ask? Some genes when inactive do not produce a measurable phenotypic change or perhaps one doesn’t know where to look or what to measure. Or there might be more than one gene capable of performing the same function (genetic redundancy66) and thus inactivating one gene may not lead to a phenotypic change because another similar gene is still functional.
7.4 Homework
© 2019, Maria Gallegos. All rights reserved.