Chapter 9 TWAS
9.1 TWAS data prep
Now that we’ve completed a traditional GWAS analysis, lets run a Transcription Wide Association Study (TWAS). That is, testing the following hypothesis: Ho: No association between gene expression and trait. Ha: There is an association between gene expression and trait.
First we need to prepare the expression matrix. The expression data comes from the same 942 maize inbred lines used in the GWAS portion of the workshop (Mazaheri et al. 2019). Raw RNAseq reads were processed as described in (Zhou et al. 2020) to generate a transcripts per million (TPM) expression matrix for ~46,430 gene models in maize.
We will demonstrate how to create a subset of 501 genes sampled equally from the 10 maize chromosomes. The phenotypes used in this section are the same simulated phenotypes used from the GWAS section.
Packages used in this section are: (Dowle and Srinivasan 2021), tidyverse (Wickham 2021), rtracklayer (Lawrence, Carey, and Gentleman 2021), rrBLUP (Endelman 2019).
Load nececarry packages
library(data.table)
library(tidyverse)
library(rtracklayer)
library(scales)Read in 942 expression matrix generated
setwd("~/PGRP_mapping_workshop/data/expression_data/")
expr = fread("widiv942_TPM.tsv", data.table = FALSE)Keep only protein coding genes gff file
setwd("~/PGRP_mapping_workshop/data/gene_annotation_data/")
gtf <- rtracklayer::import('Zea_mays.B73_RefGen_v4.48.chr.gff3')
gtf_df=as.data.frame(gtf)
gff_gene = gtf_df[gtf_df$type=="gene",] # Protein coding genesKeep needed columns from gff file
gff_keep_cols = c("seqnames","source","type","start","end","strand","gene_id","description","biotype")
out = gff_gene[,colnames(gff_gene)%in%gff_keep_cols]
genes_keep = out$gene_id
length(unique(genes_keep)) # 39,272 protein coding genesRead in Metadata
setwd("~/PGRP_mapping_workshop/data/expression_data/")
name_convert = fread("01.meta_widiv942.txt", data.table = FALSE)
name_convert = name_convert[,c("SampleID","Genotype")]Read in phenotypic data
setwd('~/PGRP_mapping_workshop/data/genotypic_and_phenotypic_data/')
phenos = fread("WIDIV_942.phe")Add a row for gid to metadata
name_convert = rbind(c("gid","gid"),name_convert)Gene coordinates of expression matrix
gene_loc = out[,c("gene_id","seqnames","start","end")]
colnames(gene_loc) = c("geneid","chr","s1","s2")Keep only protein coding genes from .gff file
expr_sub = expr[expr$gid%in%gene_loc$geneid,]Change sample names in columns to name in phenotyps file
names(expr_sub) = plyr::mapvalues(names(expr_sub), from = name_convert$SampleID, to = name_convert$Genotype)
colnames(expr_sub)[1] = "id"
rownames(expr_sub) = expr_sub$idCheck variance across all lines for expression in each data file
variance = data.frame(apply(expr_sub[,2:ncol(expr_sub)], 1, var))
variance$names = rownames(variance)
colnames(variance) = c("Var", "Gene")Remove genes with zero variance
noVar = subset(variance, Var == 0)
noVar.genes = noVar$Gene
dim(expr_sub)
expr_sub = expr_sub[!expr_sub$id%in%noVar.genes,]
dim(expr_sub) # 38,593 genesRemove genes not expressed in at least 5% of lines
low_expr = data.frame(apply(expr_sub[,2:ncol(expr_sub)], 1, function(x){sum(x == 0)}))
low_expr$names = rownames(low_expr)
colnames(low_expr) = c("Low_expr", "Gene")
Low = subset(low_expr, Low_expr > (ncol(expr_sub)-1)*0.95)
Low.genes = Low$Gene
expr_sub = expr_sub[!rownames(expr_sub)%in%Low.genes,]
dim(expr_sub) # 34,498 genesMatch name order to phenotype file file
phenos_order = c("id",phenos$Taxa)
expr_sub = expr_sub[,order(match(colnames(expr_sub),phenos_order))]
identical(as.character(colnames(expr_sub)[2:ncol(expr_sub)]), as.character(phenos$Taxa))
gene_loc = gene_loc[gene_loc$geneid%in%expr_sub$id,]Make quantile rank normalized gene expression matrix
mat = expr_sub[,2:ncol(expr_sub)]
dim(mat)
mat = t(apply(mat, 1, rank, ties.method = "average",na.last = "keep"))
mat = qnorm(mat /ncol(expr_sub))Scale TMP values to be between -1 and 1 to be compatible with rrBLUP
mat = t(apply(mat, 1, rescale, to = c(-1,1)))
mat = cbind(expr_sub$id, mat)
colnames(mat)[1] = "id"Make output file
mat_out = left_join(gene_loc,mat, by = c("geneid"="id"), copy = TRUE)Keep only genes on chromosomes, remove mitochondrial and chrolorplast genes
chrm_keep = as.factor(seq(1,10,1))
mat_out = mat_out[mat_out$chr%in%chrm_keep,]
dim(mat_out) # 34,355 genesSample 50 gene models per chromsome and make sure to include P1
set.seed(123)
mat_sub = mat_out %>%
group_by(chr) %>%
sample_n(50)See if P1 is in the filtered expression matrix
is_p1 = mat_sub[mat_sub$geneid=="Zm00001d028854",]
dim(is_p1) # Not in the subset dataBind P1 expression to the matrix
p1 = mat_out[mat_out$geneid=="Zm00001d028854",]
mat_sub = bind_rows(p1,mat_sub)Sort by chromosome and transcription start site from gff file
mat_sub = mat_sub %>%
arrange(chr,s1)Write the file
setwd("~/PGRP_mapping_workshop/data/expression_data/")
fwrite(mat_sub, file = "subset_widiv942_scaled_quant_rank_TPM.txt",col.names = TRUE, sep = "\t", quote = FALSE)Look at histogram of expression of P1
hist(as.numeric(p1[,5:ncol(p1)]))9.2 TWAS analysis
Load nececarry packages
library(data.table)
library(rrBLUP)
library(tidyverse)
library(rMVP)Read in phenotypes
setwd("~/PGRP_mapping_workshop/data/genotypic_and_phenotypic_data/")
phenos = fread("WIDIV_942.phe", data.table = FALSE)Read in expression data and put in the format for rrBLUP
setwd("~/PGRP_mapping_workshop/data/expression_data/")
expr = fread("subset_widiv942_scaled_quant_rank_TPM.txt", data.table = FALSE)
expr = expr[,-4]
colnames(expr)[1:3] = c("marker","chrom","pos")Read A matrix
setwd("~/PGRP_mapping_workshop/data/genotypic_and_phenotypic_data/")
A.widiv = attach.big.matrix("WIDIV_942.kin.desc")
A.widiv = A.widiv[,]
mean(diag(A.widiv)) # = 2 because inbred linesCheck files are in the same order
identical(as.character(phenos$Taxa), as.character(colnames(expr[,4:ncol(expr)])))Run rrBLUP for each trait and write out results
setwd("~/PGRP_mapping_workshop/twas_results/")
pheno_names = colnames(phenos)[2:3]K only model
for(i in pheno_names){
fitlist = list()
fitlist[[i]] = GWAS(pheno = phenos[,c("Taxa",i)], geno = expr, K=A.widiv, min.MAF=0.0, n.core=10, P3D=TRUE, plot=TRUE)
tmp = fitlist[[i]]
colnames(tmp) = c("geneid","chrom","pos","p_value_log10")
tmp$trait = i
fwrite(tmp, file = paste("rrBLUP_TWAS_K",i,".txt",sep = ""),
col.names = TRUE, sep = "\t", quote = FALSE, na = "NA")
rm(fitlist)
}K and SNP calculated PCs from K matrix in model
for(i in pheno_names){
fitlist = list()
fitlist[[i]] = GWAS(pheno = phenos[,c("Taxa",i)], geno = expr, n.PC = 3, K=A.widiv, min.MAF=0.0, n.core=10, P3D=TRUE, plot=TRUE)
tmp = fitlist[[i]]
colnames(tmp) = c("geneid","chrom","pos","p_value_log10")
tmp$trait = i
fwrite(tmp, file = paste("rrBLUP_TWAS_KandQ",i,".txt",sep = ""),
col.names = TRUE, sep = "\t", quote = FALSE, na = "NA")
rm(fitlist)
}TWAS manhattan plot for Q and K model for high heritability trait
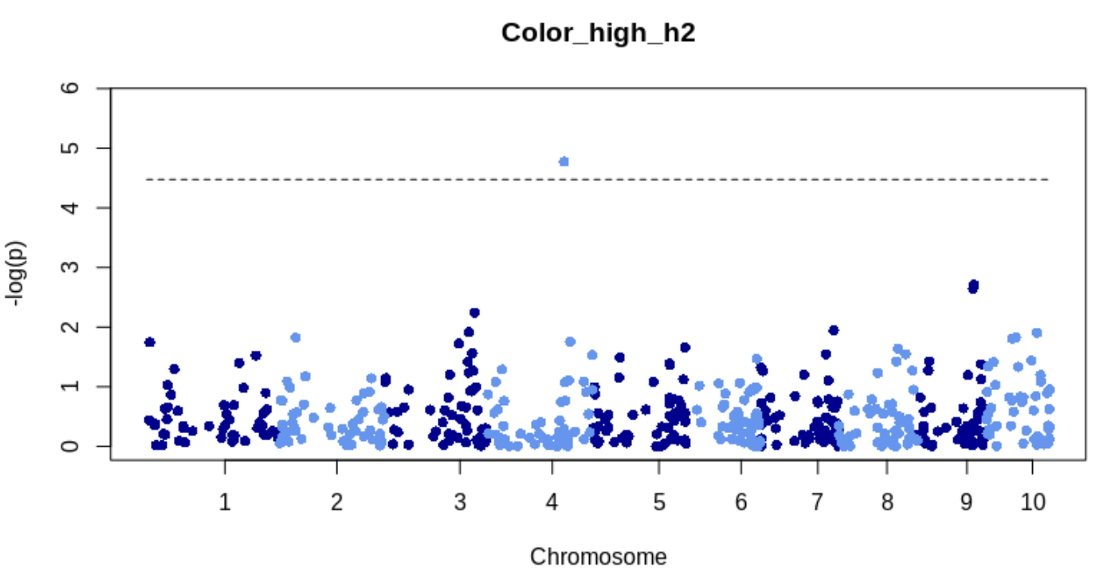
Read in results for Q and K model and make a scatterplot for most significant gene
k_q_model = fread("rrBLUP_TWAS_KandQColor_high_h2.txt")
sig_gene = top_n(k_q_model, 1, p_value_log10)
sig_gene_expr = expr[expr$marker==sig_gene$geneid,]
sig_gene_expr = data.frame(t(sig_gene_expr[,-c(2:3)]))
colnames(sig_gene_expr) = sig_gene_expr[1,1]
sig_gene_expr$Taxa = rownames(sig_gene_expr)
sig_gene_expr = sig_gene_expr[-1,]
plot_data = left_join(phenos, sig_gene_expr)
plot_data$Zm00001d052153 = as.numeric(plot_data$Zm00001d052153)Plot it
sig_plot = ggplot(plot_data, aes(Zm00001d052153, Color_high_h2)) +
# geom_jitter(colour = "darkorange",alpha = 0.3, width = 0.02) +
# geom_boxplot(alpha = 0.5, fill = "plum3") +
geom_point(alpha = 0.5, fill = "plum3")+
#ggtitle(paste0("BAF60.21 expression", " vs ", top_cis_snp)) +
geom_smooth(method = 'lm',col="darkred", aes(group=1), se=TRUE)+
theme_classic(base_size = 18)+
ylab("Trait")+
xlab("Scaled expression (TPM) Zm00001d052153") +
theme(plot.title = element_text(hjust = 0.5))
setwd("~/PGRP_mapping_workshop/figures/twas/")
pdf("WIDIV_942_signif_TWAS.pdf", width = 6, height = 4)
sig_plot
dev.off()Now do the same for P1
p1_gene_expr = expr[expr$marker=="Zm00001d028854",]
p1_gene_expr = data.frame(t(p1_gene_expr[,-c(2:3)]))
colnames(p1_gene_expr) = p1_gene_expr[1,1]
p1_gene_expr$Taxa = rownames(p1_gene_expr)
p1_gene_expr = p1_gene_expr[-1,]
plot_data1 = left_join(phenos, p1_gene_expr)
plot_data1$Zm00001d028854 = as.numeric(plot_data1$Zm00001d028854)Plot it
p1_plot = ggplot(plot_data1, aes(Zm00001d028854, Color_high_h2)) +
geom_point(alpha = 0.5, fill = "plum3")+
geom_smooth(method = 'lm',col="darkred", aes(group=1), se=TRUE)+
theme_classic(base_size = 18)+
ylab("Trait")+
xlab("Scaled expression (TPM) P1") +
theme(plot.title = element_text(hjust = 0.5))
pdf("WIDIV_942_signif_TWAS_P1.pdf", width = 6, height = 4)
p1_plot
dev.off()