Chapter 4 Quality control
4.1 FastQC
Quality control (QC) is fundamental and is involved in all RNA-Seq pre-processing. After sequencing the reads, you need to check the quality of the sequencing and FastQC does this very well. FastQC presents in a simple way some checks control on raw sequence data resulting from high-throughput sequencing. This tool shows the problems that exist in the data, problems that can result in misinterpretations of the biological result. Many works have used FastQC as it accepts SAM or BAM alignment files or raw files such as FASTQ. In addition, it provides the results in graphical form of the main sequencing metrics.
FastQC can be run with a graphical interface, but we will run it in the terminal. For didactic purposes, we will work with the dummy samples that are in 0-samples:
ls ~/PreProcSEQ-main/0-samplesWe are going to use FastQC to generate the quality reports for each FASTQ file. In this case, the samples are in 0-samples and the FastQC result, we will save in 1-qualityControl_beforeTrimming/outputFastqc:
fastqc ~/PreProcSEQ-main/0-samples/*.fastq -o ~/PreProcSEQ-main/1-qualityControl_beforeTrimming/outputFastqcYou can verify that output files were generated for each FASTQ file:
ls ~/PreProcSEQ-main/1-qualityControl_beforeTrimming/outputFastqcThe .zip file contains the information and metrics used for QC. The .html file covers QC graphically:
firefox ~/PreProcSEQ-main/1-qualityControl_beforeTrimming/outputFastqc/sample_01_R1_fastqc.html4.2 MultiQC
MultiQC aggregates bioinformatics analysis results from many samples into a single report. This is what we are going to do with the data generated earlier with FastQC. Let’s set the files generated by FastQC as input. The MultiQC result will be stored in 1-qualityControl_beforeTrimming/outputMultiqc:
multiqc ~/PreProcSEQ-main/1-qualityControl_beforeTrimming/outputFastqc/. -o ~/PreProcSEQ-main/1-qualityControl_beforeTrimming/outputMultiqcThe result of MultiQC is in 1-qualityControl_beforeTrimming/outputMultiqc:
ls ~/PreProcSEQ-main/1-qualityControl_beforeTrimming/outputMultiqcWe can view the HTML file in a web browser:
firefox ~/PreProcSEQ-main/1-qualityControl_beforeTrimming/outputMultiqc/multiqc_report.htmlFigure 1 shows the main graphic results of QC generated by FastQC with MultiQC. Sequencing was generally good (Figure 1.A), all read positions will have average phred quality scores above 30. The average quality of each read was also good (Figure 1.B), some reads had averages of phred scores that were in the yellow and red region, but most were in the green region. As for the content of unidentified bases (N), the content was low, but there were Ns in the readings (Figure 1.C). Finally, adapters were found in the analyzed dataset (Figure 1.D). However, the content was not enough to be in the yellow or red region.
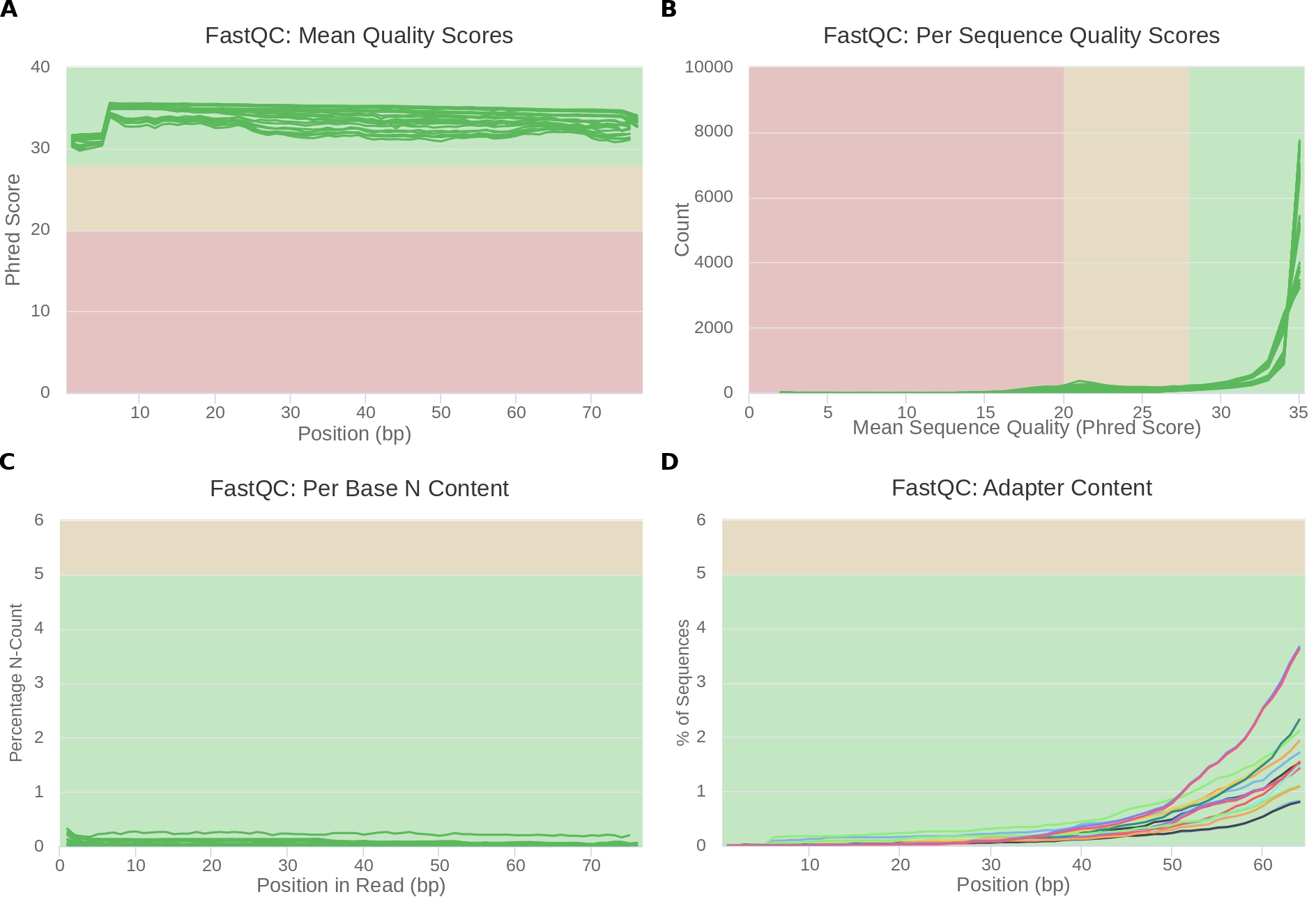
Figure 1: Quality control. The background color indicates whether the region is bad (red), acceptable (yellow), and great (green). In (A), (B) and (C) each line represents a FASTQ file of the project. (A) Average of quality scores. The X axis represents the position of the nucleotides and the Y axis indicates the quality score on the phred scale. (B) Quality score by sequence. The X axis represents the average phred score and the Y axis indicates the number of readings. (C) N content. The X axis represents the position of the base and the Y axis indicates the amount of N in percentage. (D) Content of adapters. Each line represents an adapter type.