Chapter 3 Workshop 2: Sequence alignment and prediction of structure and function
In this session, we will explore the concepts of sequence similarity and sequence alignment and how they can be used to predict the structure and function of a protein.
3.1 Using WWW resources to discover a new gene
A huge volume and range of biological data are available via the World Wide Web (WWW). In this course, we are mainly concerned with macromolecular sequence data, in other words: nucleotide sequences of genomic DNA, transcripts and amino acid sequences of proteins. These data are the product of decades of research and countless millions of £s of investment; this represents an invaluable yet freely available resource for clinicians, biotechnologists, academic researchers and students to make novel discoveries.
One of the things you will do today is to discover and characterise a novel gene! You will use some basic bioinformatics resources such as the BLAST software and sequence databases to make your novel discovery. You can work individually or in small groups, if you prefer. Try to answer the questions in the boxes below (don’t worry! This will not be assessed).
3.2 The data
A good place to access DNA sequence data is the NCBI Nucleotide database (https://www.ncbi.nlm.nih.gov/nuccore/). You can do simple (as well as quite sophisticated) searches for sequence data here. But perhaps the most useful way to access sequence data is via a BLAST search. You might already be familiar with BLAST from previous modules.
“The Basic Local Alignment Search Tool (BLAST) finds regions of local similarity between sequences. The program compares nucleotide or protein sequences to sequence databases and calculates the statistical significance of matches. BLAST can be used to infer functional and evolutionary relationships between sequences as well as help identify members of gene families.”
3.3 Discover a previously unknown gene
The data in this Nucleotide database include complete chromosomes, individual genes, expressed sequence tags (ESTs) and various other DNA/RNA molecules and fragments of molecules. Some of these DNA sequences have been very well described and annotated, e.g. sequences of human chromosomes. However, many DNA sequences in the database have hardly been studied at all and present an opportunity for us to discover something new. Examples include some microbial genome sequences and a wealth of metagenomics sequences. These are a good place to go ‘fishing’ for new genes. In fact there is a name for this: ‘bioprospecting.’

A genome (or metagenome) sequence typically contains segments called genes that encode a protein (or a functional RNA), separated by intergenic regions. Generally, the genes evolve relatively slowly, so they will share sequence similarity with genes from other organisms, even if that similarity is limited. On the other hand, intergenic regions often evolve relatively quickly and may share little or no sequence similarity with any other organisms. So, we are going to look for islands of relatively conserved sequence to help identify new genes.
3.3.1 Choose a previously uncharacterised DNA sequence
The first step towards discovering a new gene is to choose a DNA sequence from the public databases that has not already undergone extensive study and gene prediction. A good source of such sequences are the various metagenomics studies of microbiomes. Try searching for a suitable example at the NCBI Nucleotide web portal: https://www.ncbi.nlm.nih.gov/nucleotide/
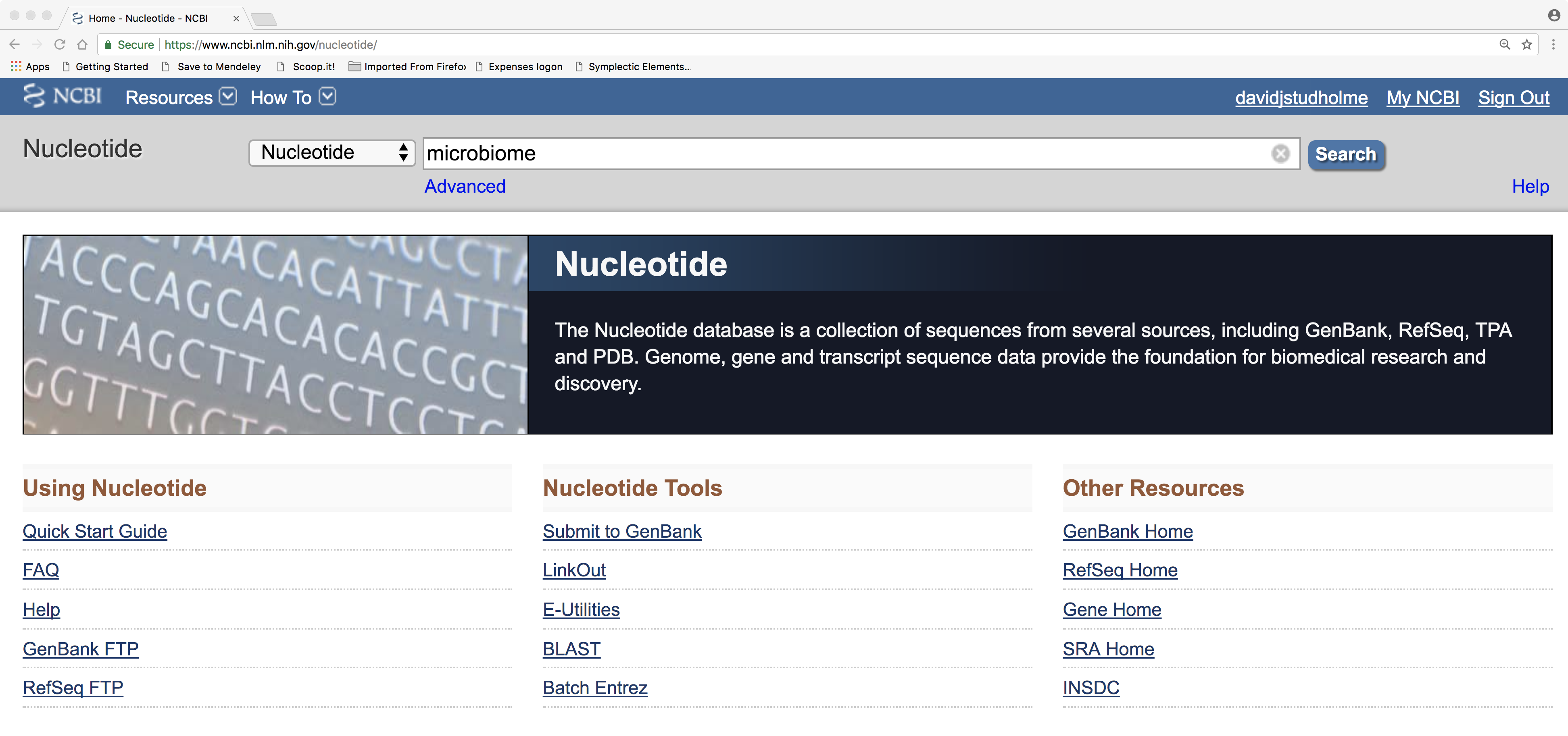
At the NCBI website you could simply search for ‘metagenome’ or something more specific like ‘coral metagenome,’ ‘alkali sediment metagenome’; use your imagination! Then choose a specific DNA sequence from this metagenome/genome to work on, from among those returned by the search. You might need to use some trial and error to navigate around the NCBI website, clicking on a few links before you get to a specific sequence. Do spend some time trying to figure out the logic of how the data are organised in these web pages.
Do you know what a metagenome is? If not, then “Google” it and/or ask!
See an example of a candidate DNA sequence for analysis in the image below:
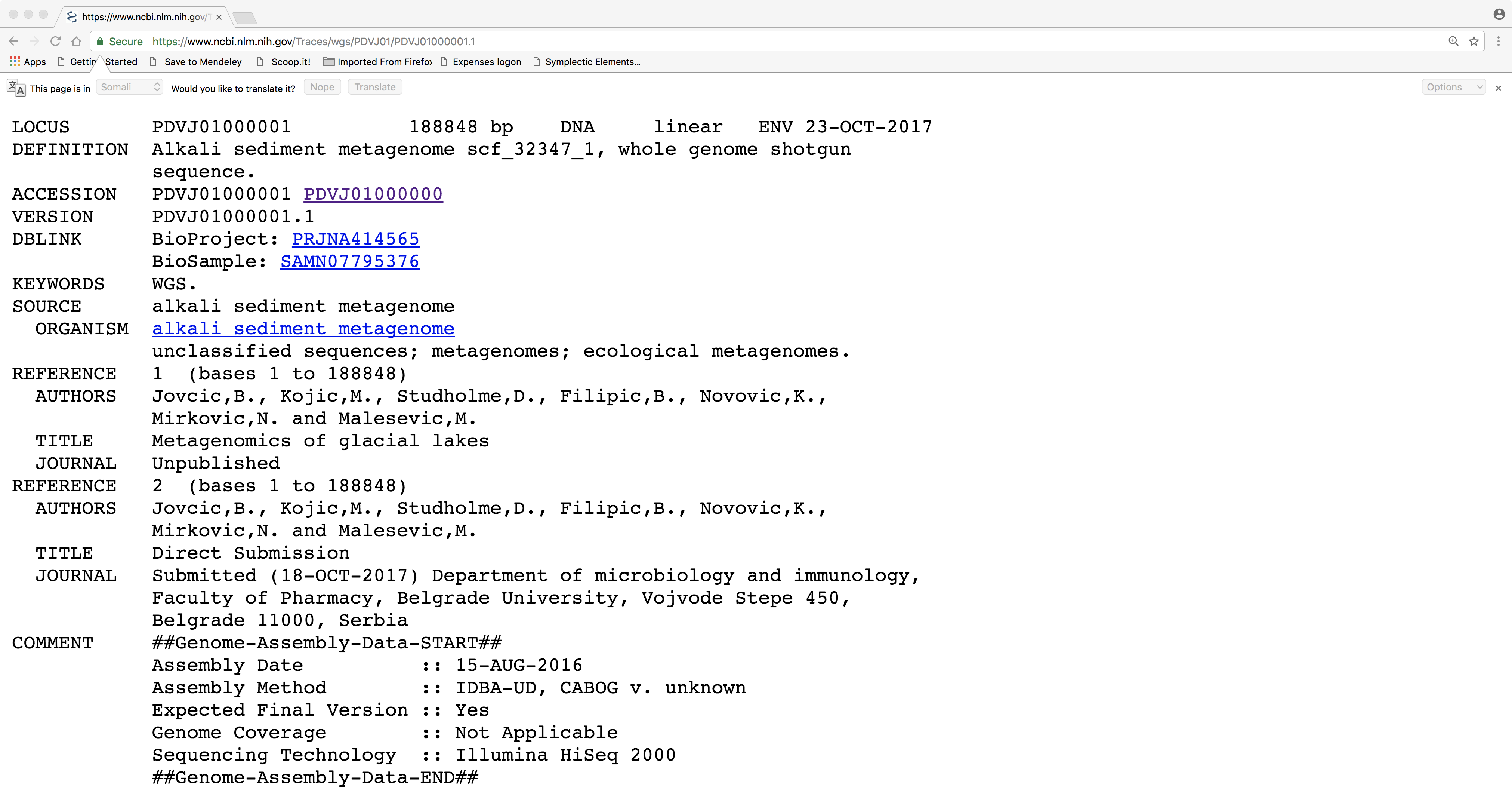
I recommend that you work through this using the illustrated example before choosing your own sequence.
The example used in the remainder of this document comes from a metagenome. However, instead you could use an unanotated genome. This would be a good idea; analysis of an unannonated microbial genome fits well with subsequent Computer Practical sessions and, maybe, the coursework assignment.
Here are some good examples that you could use:
Choose one specific DNA sequence.
For the DNA sequence that you have chosen to study, can you find the following information?
| Question | Please write you answer in this space |
|---|---|
| Where did this DNA sample come from? | |
| What method was used to sequence the DNA? | |
| Accession number | |
| How long is this DNA sequence? | |
| Do we know what organism this DNA came from? | |
| Are there any research papers associated with this DNA sequence? | |
| Who generated the data? |
Now, we are going to try to discover a new protein-coding gene on this DNA sequence. In other words, we are going to annotate the naked DNA sequence with the positions of a gene. The underlying principle is that the protein encoded by this gene shares some sequence similarity with already-known proteins. So we are going to search for patches of sequence similarity between our naked DNA sequence and a database of amino-acid sequences from known proteins.
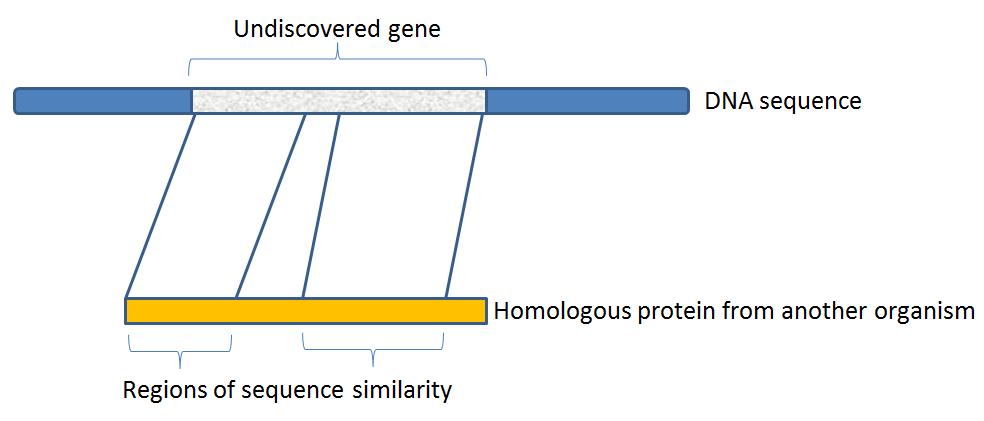
To acheive this, we will perform a BLAST search using your chosen DNA sequence as the query against the databases of protein amino acid sequences at the NCBI. Remember, there are several flavours of BLAST (BLASTN, BLASTP, TBLASTN, BLASTX, TBLASTX); which one should you use and why? (Clue: BLASTX takes a DNA query sequence and searches against a database of proteins). You can perform your BLAST search at https://blast.ncbi.nlm.nih.gov/Blast.cgi as in the example in the image below.
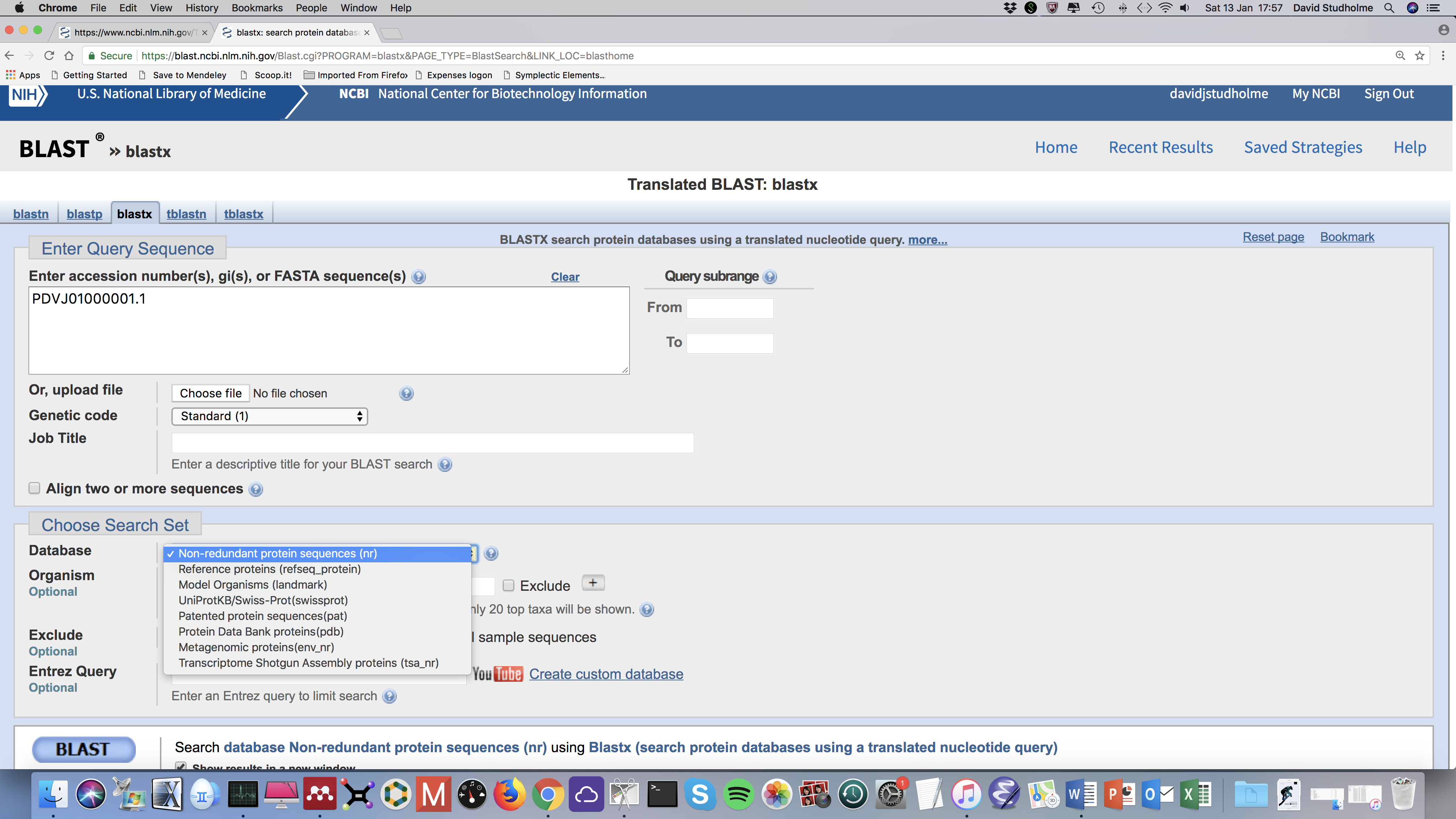
Notice that for the query you can insert either the actual DNA sequence or just the accession number. When you are ready, hit the “BLAST” button and wait for the results. By the way, if your patience is limited, you might wish to search against the Swiss-Prot database rather than the default database because searching this smaller database will be a bit quicker than searching the much larger one. On the other hand, searching against the smaller, less-comprehensive database might yield less information. So, you could try both simultaneously (in different tabs in your web browser).
In the example BLAST result below, we can see four regions of the DNA sequence that show clear similarity. This strongly suggests there is homology between the product of our query sequence and previously described proteins in the SwissProt database. Since these previously-known proteins are homologous to our query protein, it is likely that they share similar structure and function.
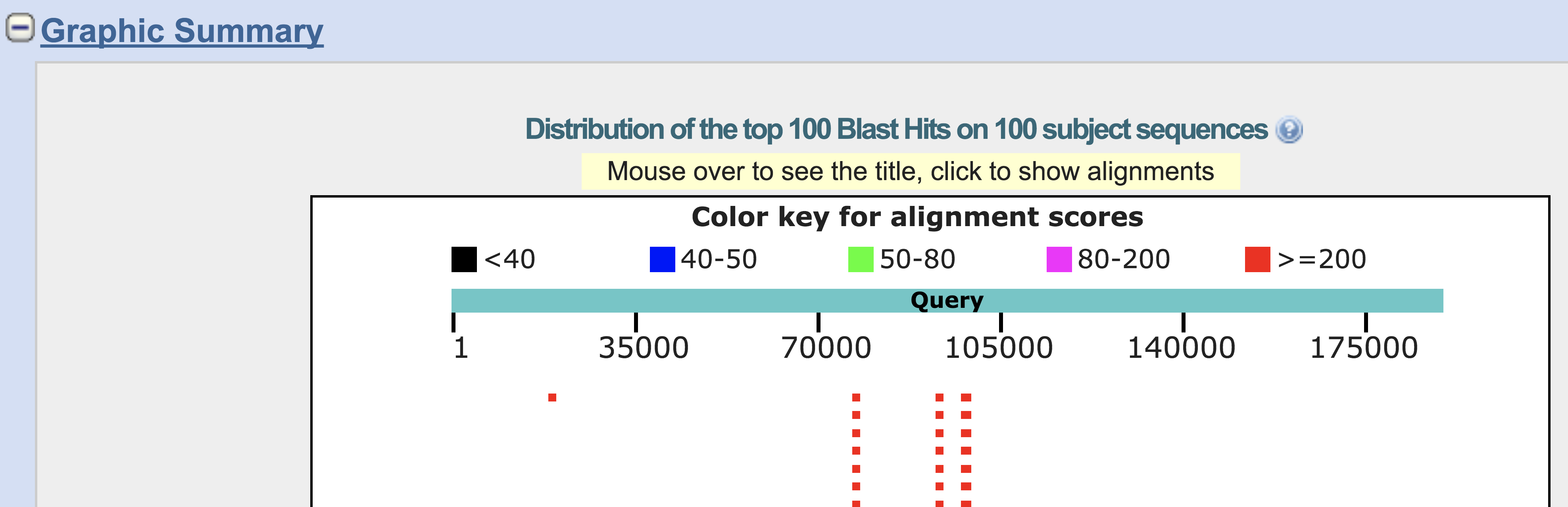
Let’s choose one of these for further investigation (click on one of the four red blocks that each represent a block of sequence similarity):
We are now going to perform a series of steps in order to extract the DNA sequence of our newly discovered gene. Note that this gene sequence is a substring of the original query DNA sequence that we started with. In other words, the protein-coding gene sequence occupies only a small part of the genomic DNA sequence. In the example above, our gene lies around positions 76,476 and 77,696 in the original 188,848-bp sequence. There is more than one way to do this. For example, we could try to manually copy and paste this substring onto the clipboard. However, the steps below are probably easier and less error-prone.
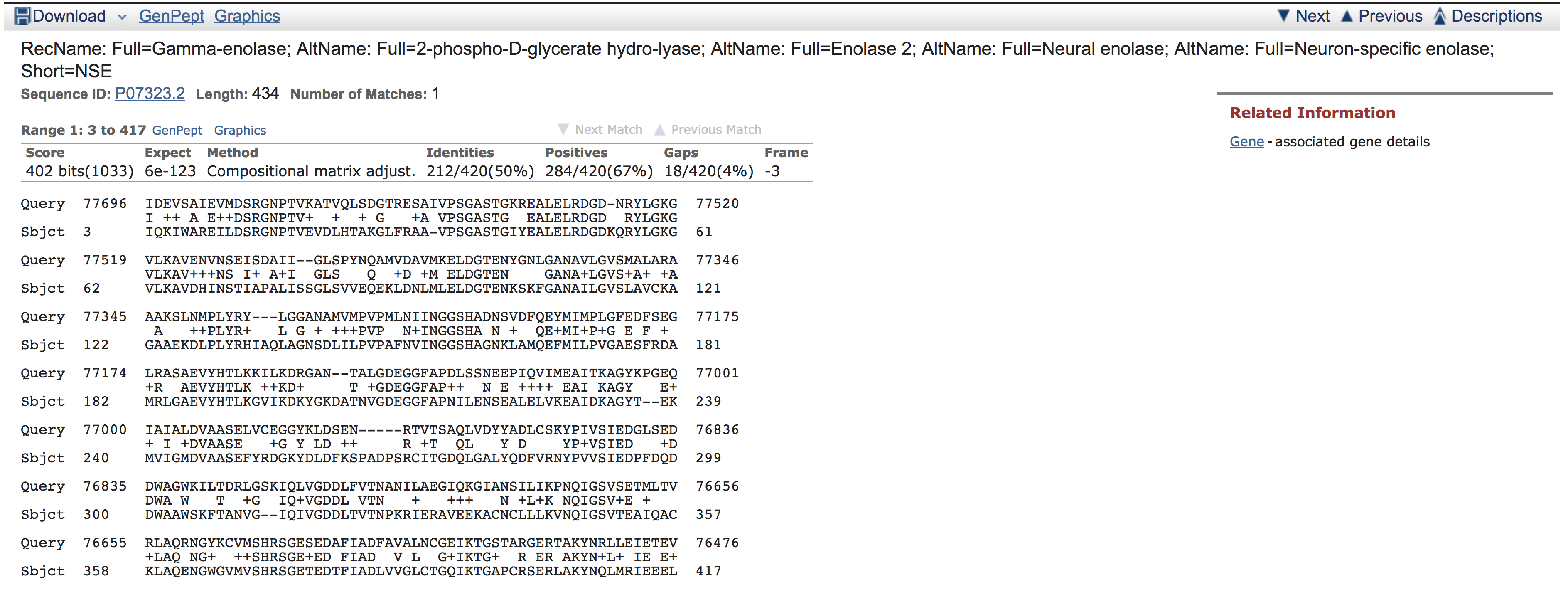
In the example above, we can see that protein accession P07323.2 is homologous with our new gene. So, let’s use TBLASTN to get the sequence of our new gene by aligning the protein against the DNA sequence. This time we will use the “Align two or more sequences” option and we will use previously described protein P07323.2 as the query and metagenomic DNA sequence accession PDVJ01000001.1 as the ‘subject sequence’ against which we will search the query:
The result of this example looks like this:
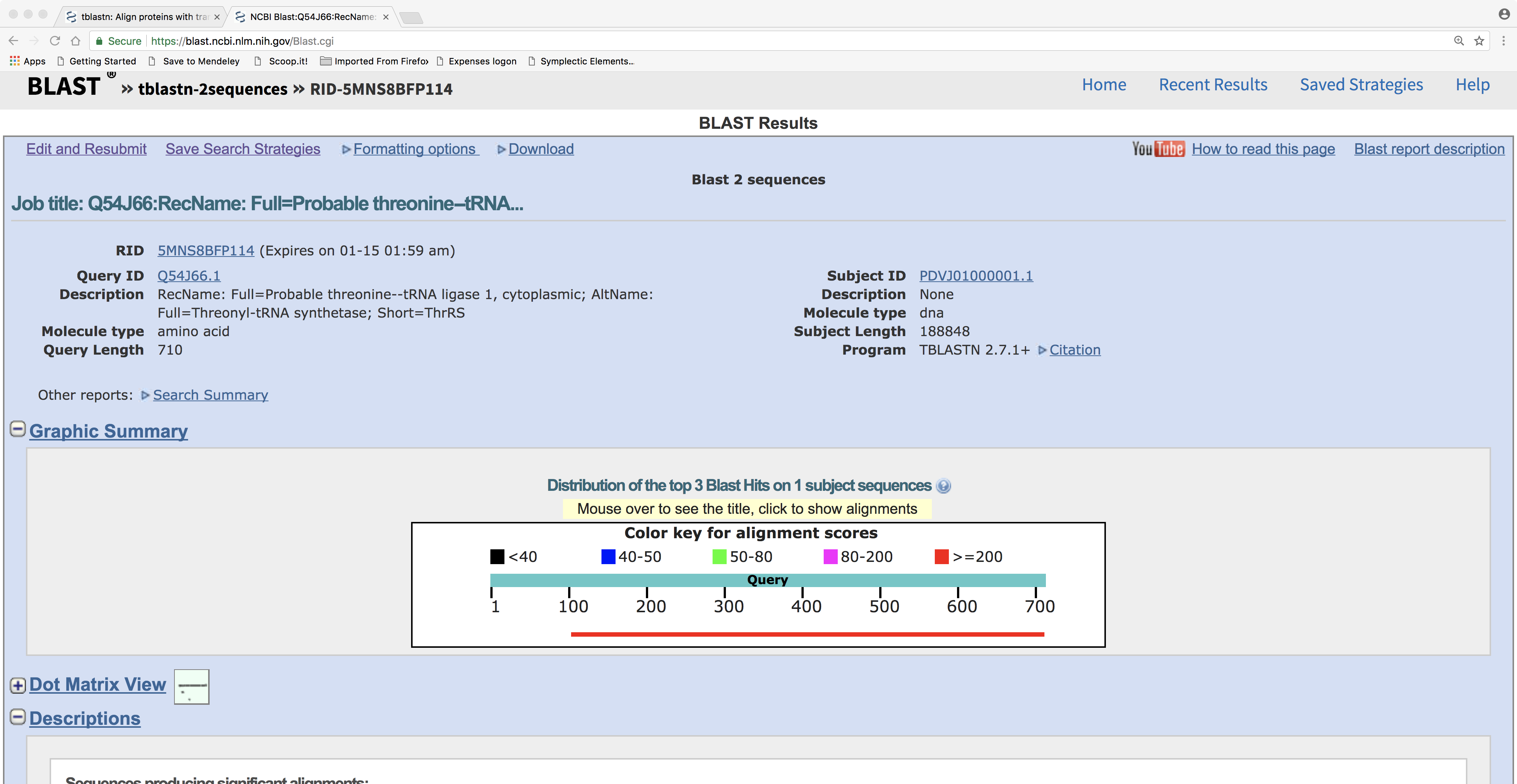
This clearly shows that about 85% of the query protein’s length has detectable sequence similarity with our query DNA sequence. Now if we scroll down we can see the sequence alignment:

Note the “Download” icon (in the figure above). This allows us to obtain the sequence of our new gene:
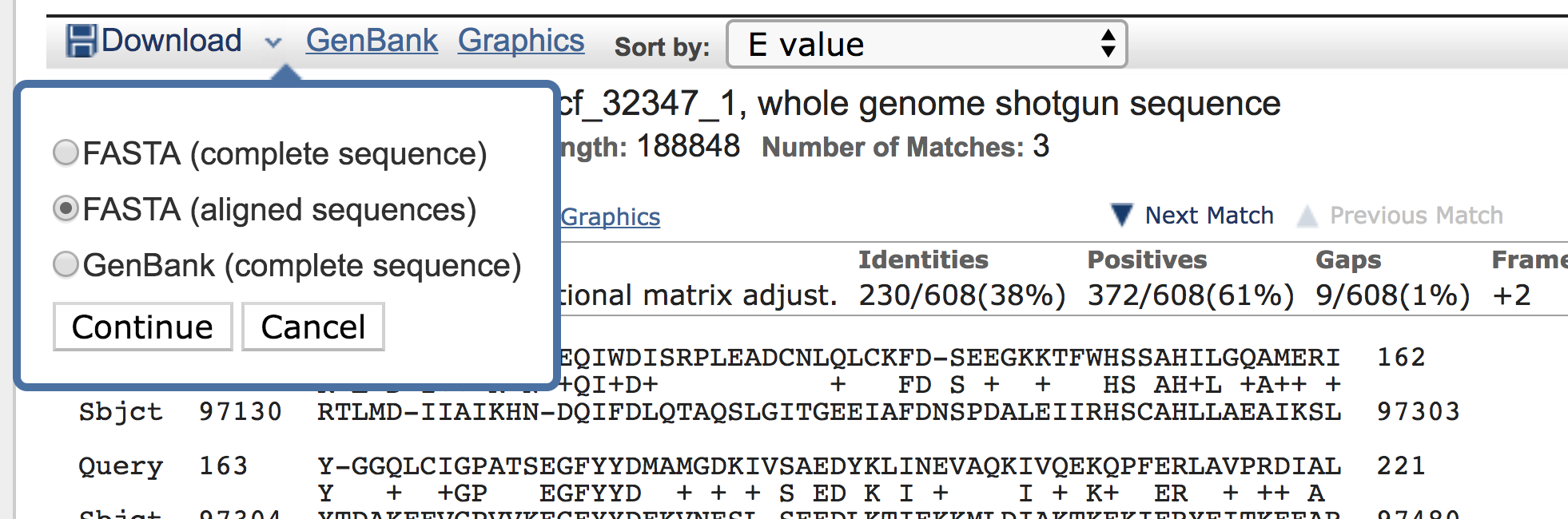
Here is the resulting nucleotide sequence of our new gene:

Notice that this contains just the sequence of the actual gene and we have discarded the non-coding DNA sequence from upstream and downstream of the gene. We can now perform analyses on this gene without confusing or confounding results arising from the up- and downstream flanking sequences.
We can now try to gain insights into the structure, function and evolution of this gene by pasting its sequence, or the amino-acid sequence of its encoded protein, into various publicly available webservers …
Once you have extracted the sequence of the gene that you have discovered, try to answer the following questions about it. Some hints are given below about online tools that you could use to address these questions. BLAST searches will help to answer many of these. The most useful thing to do first is to translate from DNA to protein. Experiment with the tools mentioned below, copying and pasting your new protein sequence. They are all reasonably easy to use; but please ask for help if you get stuck. Discuss your findings with other students, the demonstrators and lecturer.
| Question | Please write you answer in this space (it may be useful for your coursework) |
|---|---|
| What is the predicted function? | |
| Does it have any significance in pathogenesis, biotechnology, fundamental biological processes? Hint: PubMed database might be helpful. | |
| Can we predict the structure of the protein? | |
| Does it contain any structural domains? | |
| Is the gene part of an operon or cluster of functionally related genes? | |
| Do homologues occur in only closely related organisms or do they occur more widely across all domains of life? | |
| Can you generate a multiple sequence alignment for your protein and its homologues? | |
| From the alignment, can you build a phylogenetic tree? | |
| What organism (taxonomic group) does your protein come from? The phylogenetic tree might help you to answer this. |
Suggested online analysis tools (additional information about some of these is provided below):
| Suggested tool | Web address |
|---|---|
| Translate DNA to protein | https://web.expasy.org/translate/ |
| STRING (see mini-tutorial below) | https://string-db.org/ |
| Pfam | http://pfam.xfam.org/ |
| Find open reading frames in your gene sequence | http://www.ncbi.nlm.nih.gov/gorf/gorf.html |
3.4 Generating a sequence alignment and phylogenetic tree
Placing a novel protein/gene into a phylogenetic tree can reveal insights into its evolutionary origins and possibly its function. Remember that the example query sequence GenBank: PDVJ01000001.1 came from a metagenome. A metagenome is basically a mixture of many individual genomes potentially from many different species. How do we know which species this sequence originated from? A phylogenetic analysis can help here …
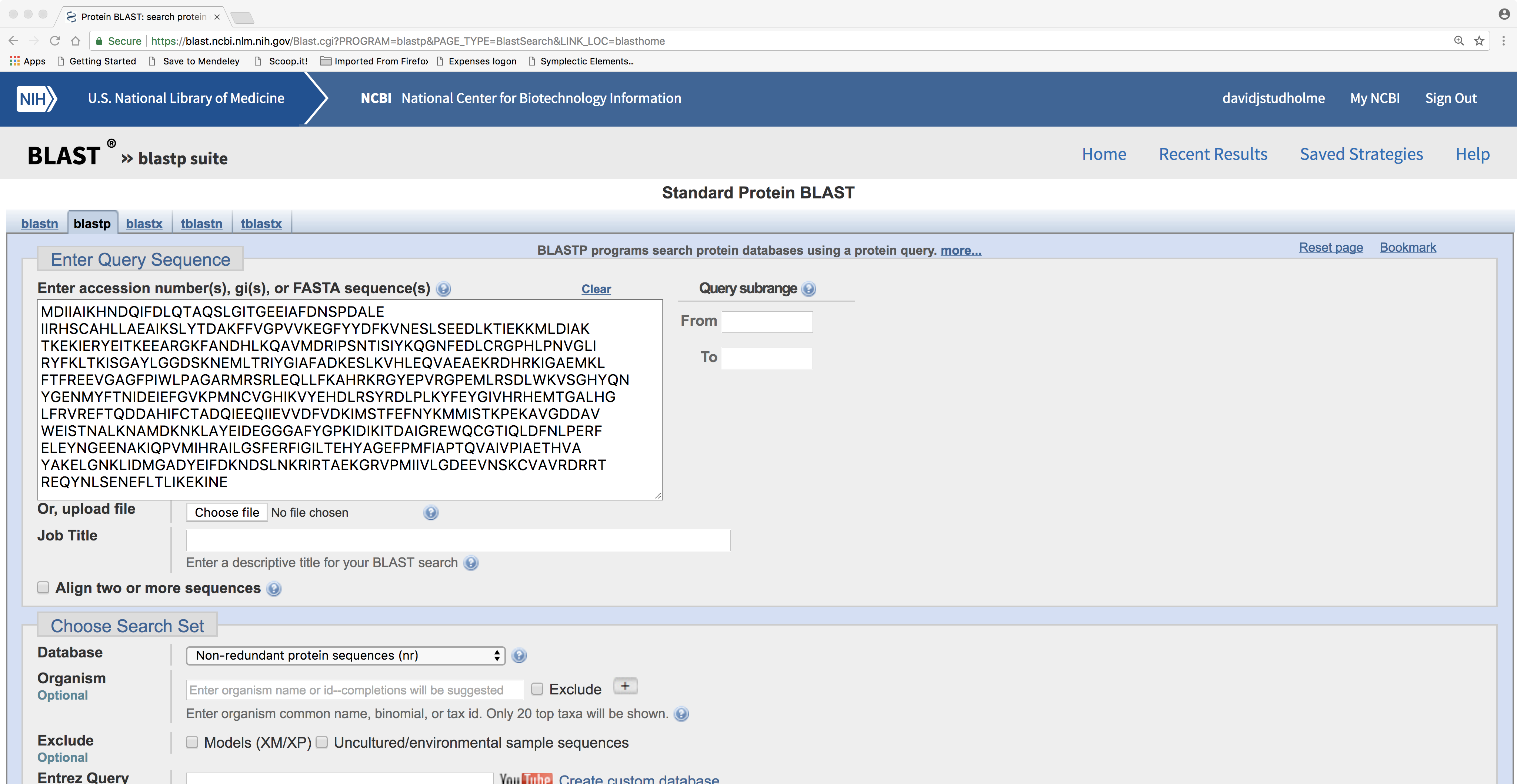
Please see the following series of screenshots illustrating how to use BLAST to gather homologues (i.e. related sequences) of our query sequence, align them and generate a phylogenetic tree.
Here is the BLASTP search form:
Here is the BLASTP result page. Note the graphical representation of protein domain structure. Also note hyperlinks to “Multiple alignment” and “Distance tree of results”:
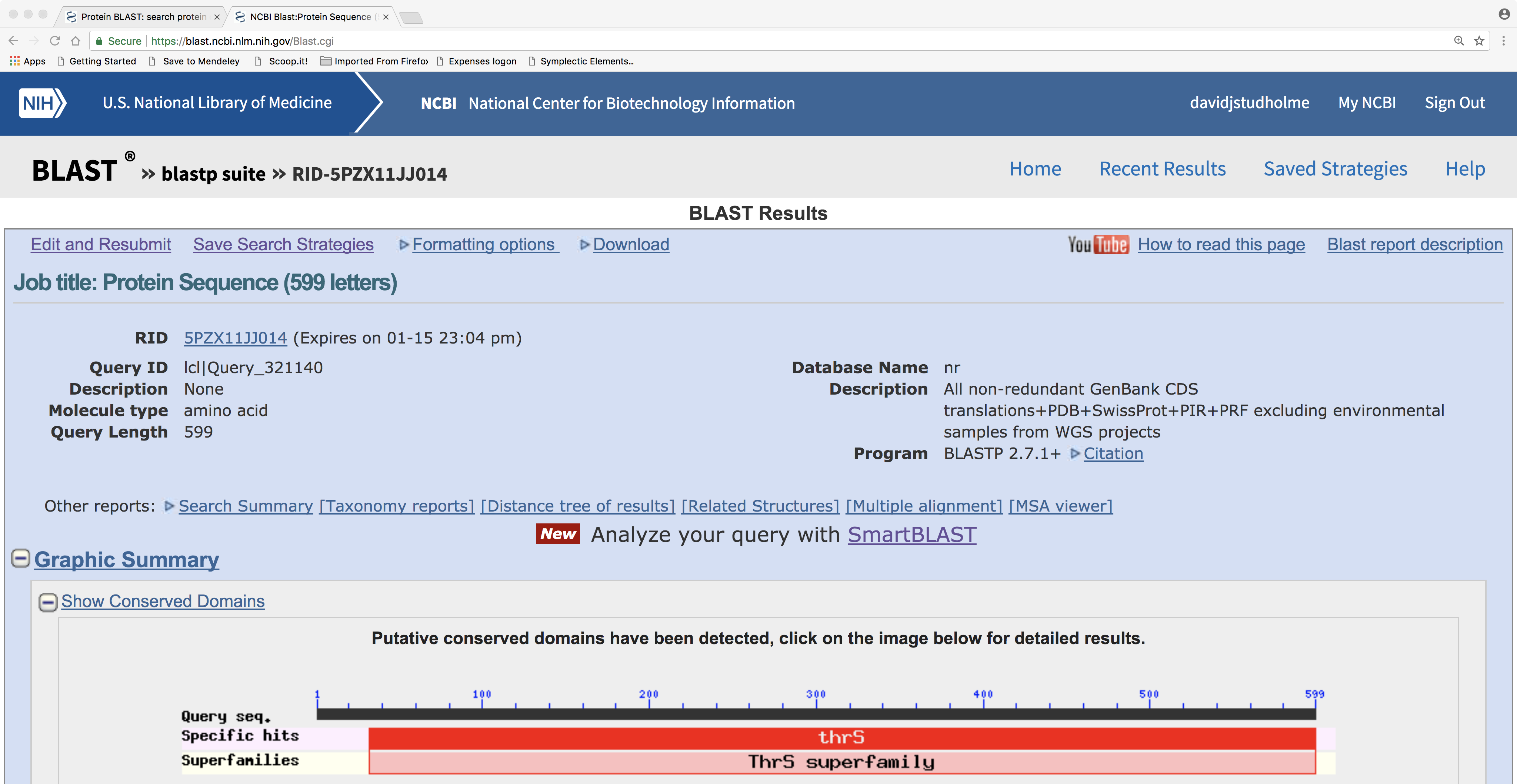
3.4.1 A note about protein domains
Often, a single polypeptide chain within a protein can be made-up of multiple domains. Each domain may have a different function; for example, a transcription factor protein might contain a DNA-binding protein combined with another domain that interacts with other regulatory proteins. That same DNA-binding domain might also occur in some other protein, perhaps containing a protein-kinase domain. So, proteins are modular, comprised of basic functional and structural building blocks that are the domains. See pages 370-371 in A. Lesk (2017).
There are several databases that allow us to recognise domains within an amino-acid sequence. These include Pfam, SMART, TIGRfams, among others. The NCBI’s BLAST web page presents the results of searches against these domain databases in addition to the actual search results. Searching agains a domain database such as Pfam is achieved not via a BLAST search but rather through the use of hidden MArkov models (HMMs). You can read more about the use of HMMs in pages 152 - 153 in A. M. Lesk (2019). Another tool that you can use for simultaneously searching against a whole collection of domain databases is InterProScan. I also recommend searching against the Pfam database, which is arguably the ‘best’ of these databases. By searching against just a single database, you can avoid some of the confusion that ensues from trying to interpret multiple overlapping results from multiple databases.
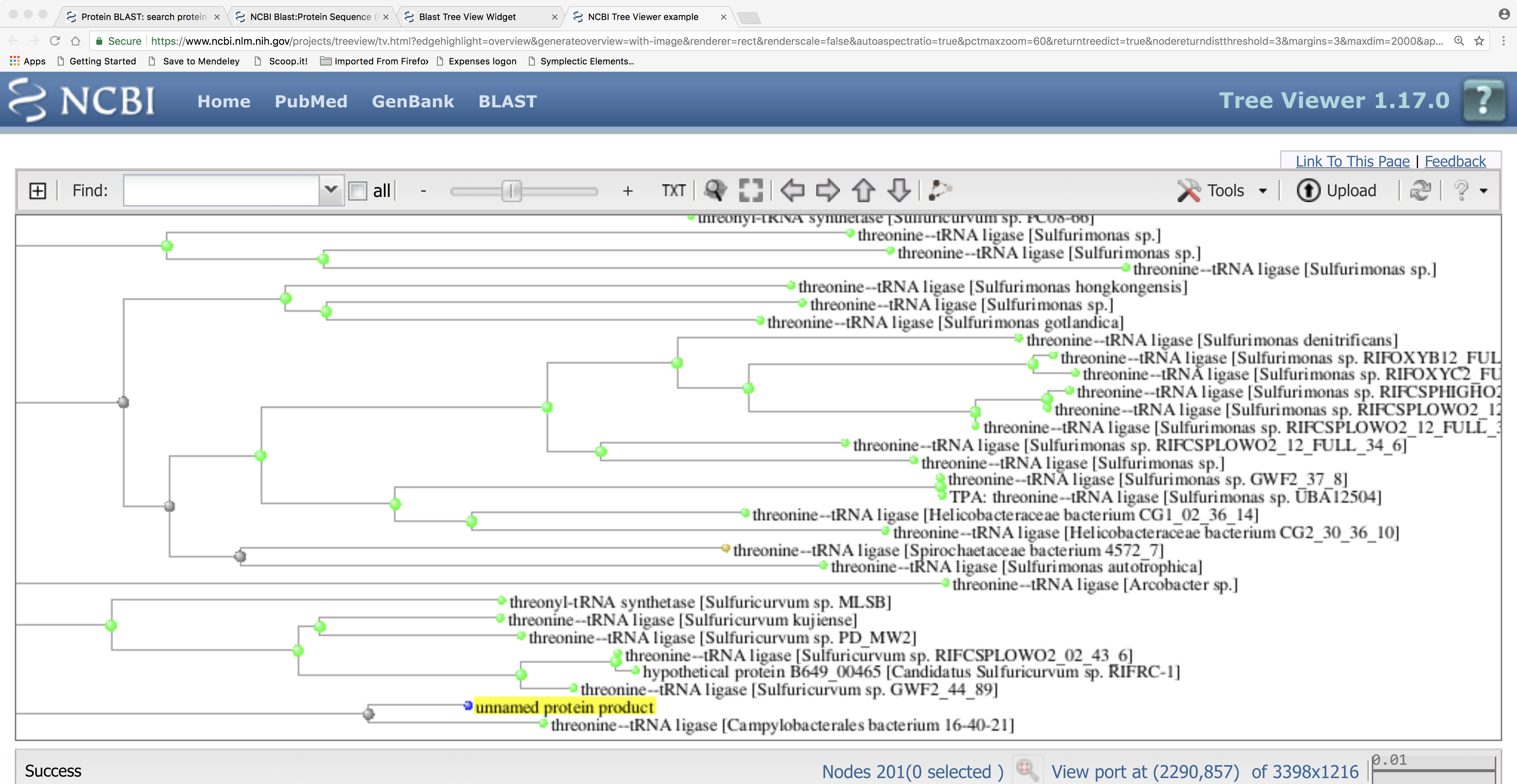
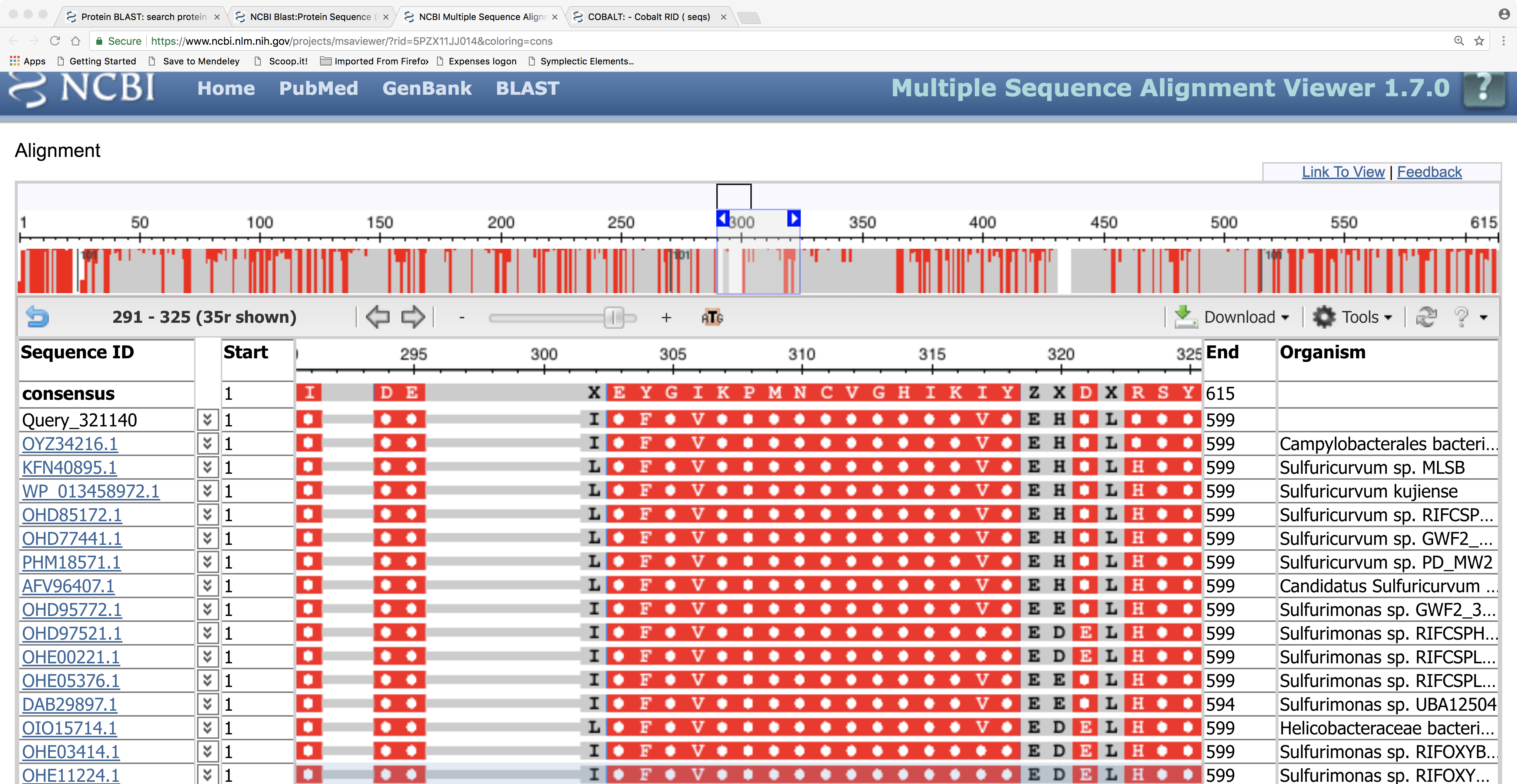
Here is the tree showing the relationships between our query protein sequence and the BLASTP hits. Note that our query protein seems to cluster with a protein from the epsilon-proteobacterial order Campylobacterales and, more distantly, to other epsilon-proteobacteria such as Sulfuricurvum spp.; this strongly suggests that this metagenomics sequence originates from a bacterium of that group. Can you think of a way to investigate or check this further, using BLAST searches?
Here we can see the details of the amino-acid sequence alignment of our query sequence against the BLASTP hits:
3.5 Predicting protein function with the STRING database (http://string.embl.de/)
Many microbial genomics databases are available that, rather than just presenting raw data, have some “added value,” such as tools for predicting the function of genes. One such example is the STRING database.
This database aims to provide clues about the functions of poorly characterised genes by identifying associations with genes of better-understood function. These associations can be physical proximity of genes in the genome or they can be correlations in patterns of presence/absence. Other associations are gleaned from mining text of published papers or looking at correlation in transcriptomics expression data. In any case, the detection of such associations depends on the interrogation of a large number of genome sequences (or transciptome and/or textual data). Let’s try it out using as an example the E. coli otsA gene, involved in trehalose biosynthesis; this example is borrowed from a previously published genomics tutorial (Strong et al., 2004). You should also try it with your newly discovered gene/protein.
First, enter the terms into the search box and press the “Search” button:
(Note that you can also search “by sequence” rather than “by name”; therefore, you could paste-in the sequence of your newly discovered protein.)
This yields a network of interactions, inferred from genome sequence data, between the otsA gene and some others:
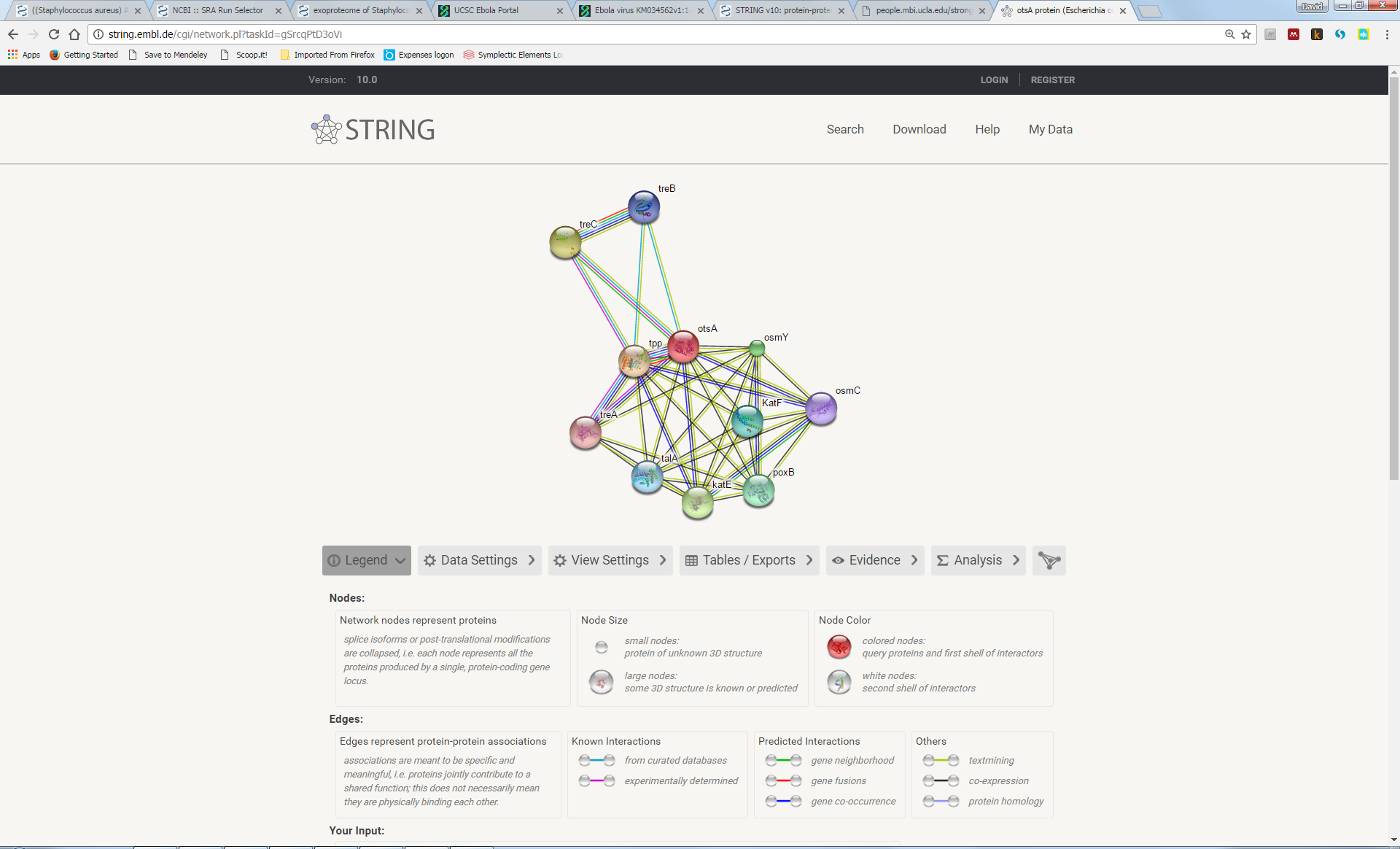
Note that this network is represented as a ‘graph’ consisting of nodes (a.k.a. ‘vertices’) and connections (‘edges’). We will encounter graphs again in other contexts of bioinformatics, especially in de-novo sequence assembly later in the course.
Note that some of these genes, associated via genome sequence data, also happen to have related functions (trehalose biosynthesis):
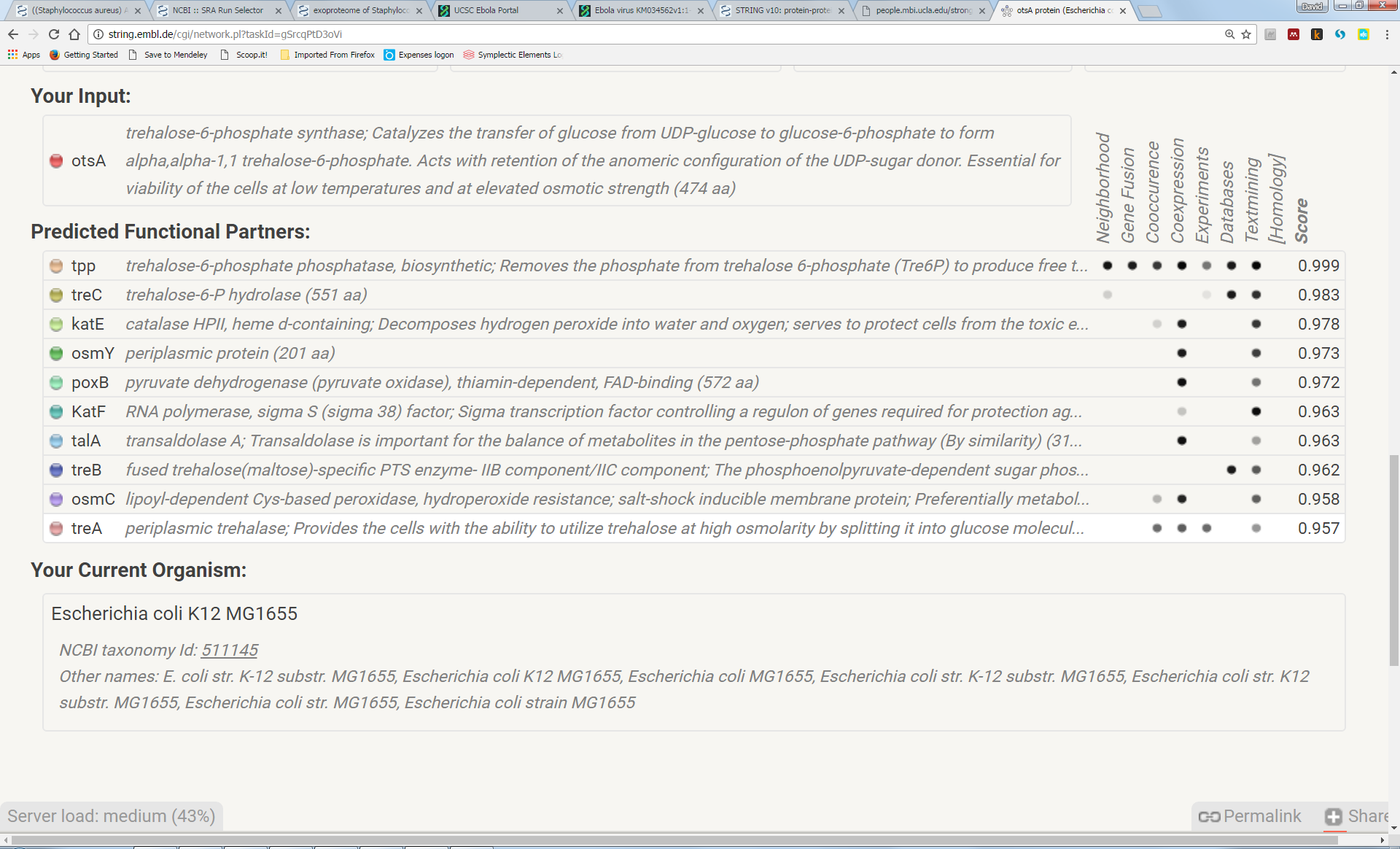
Try to understand the meaning of the realtionships that link the genes in this network. Here we can see that otsA and tpp are adjacent to each other in many genomes, suggesting a functional link (“Neighbourhood”). In a few genomes, the two proteins are actually fused into a single protein (“Gene Fusion”). Furthemore, most genomes either contain both genes or neither gene (“Cooccurrence”):
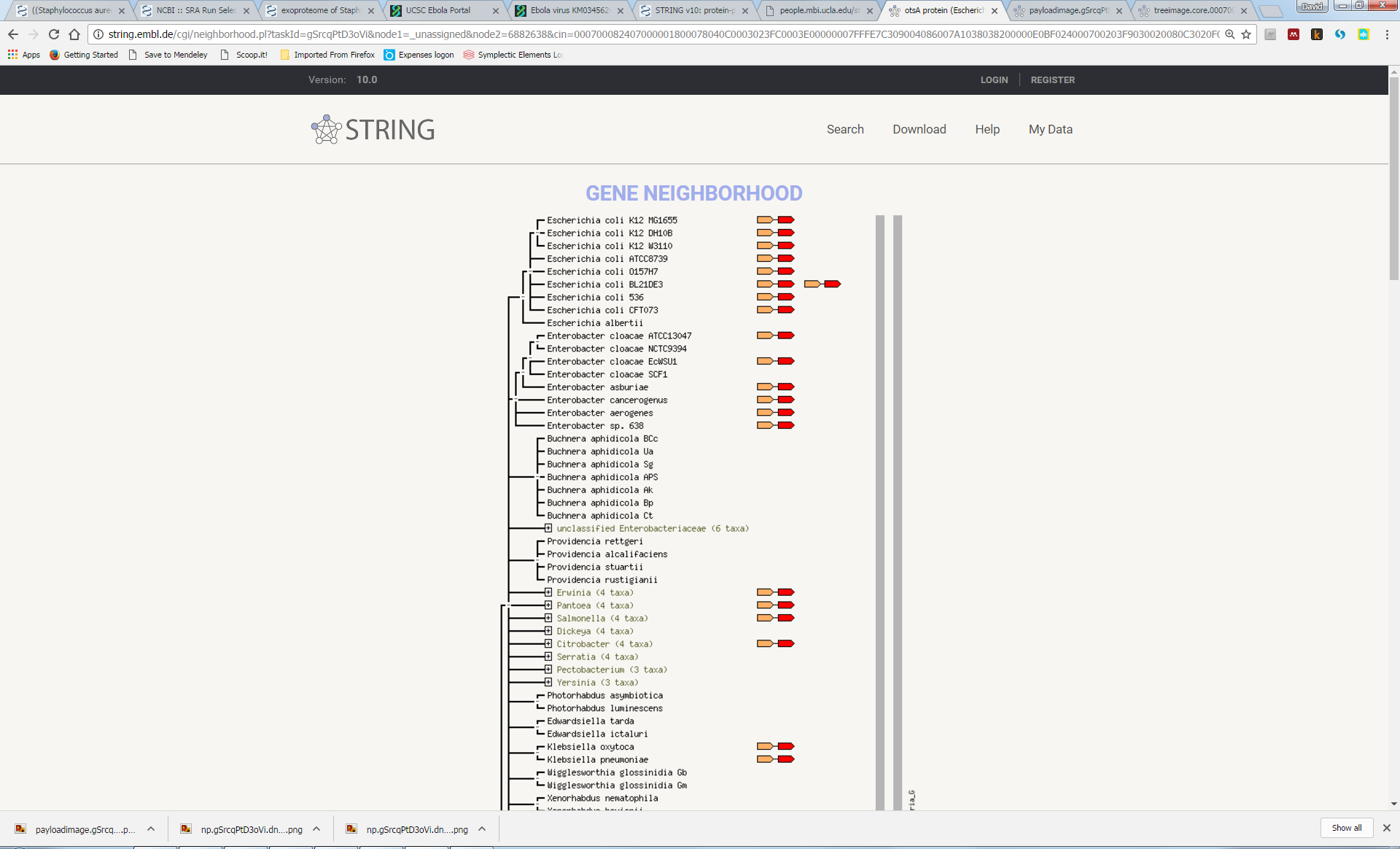
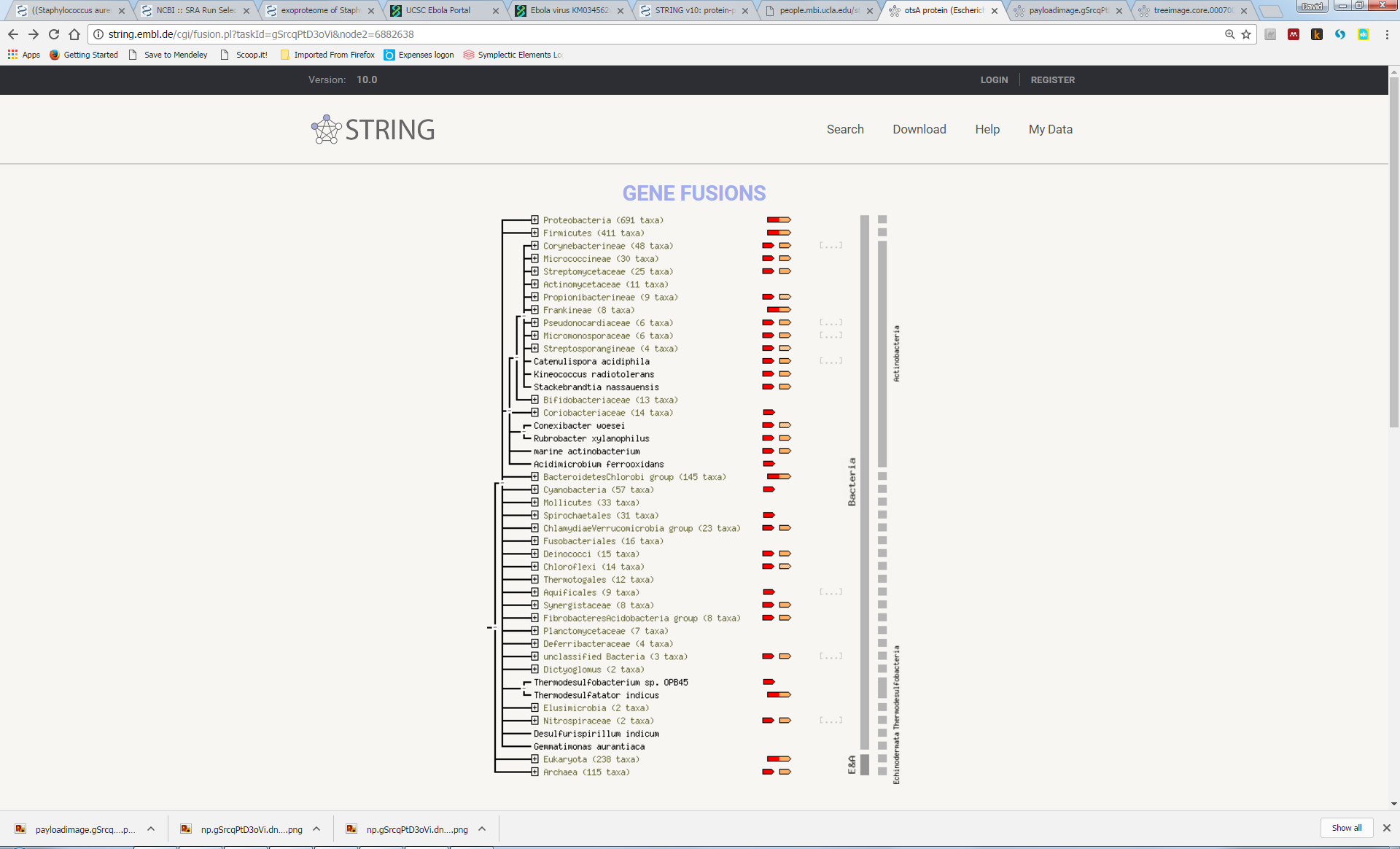
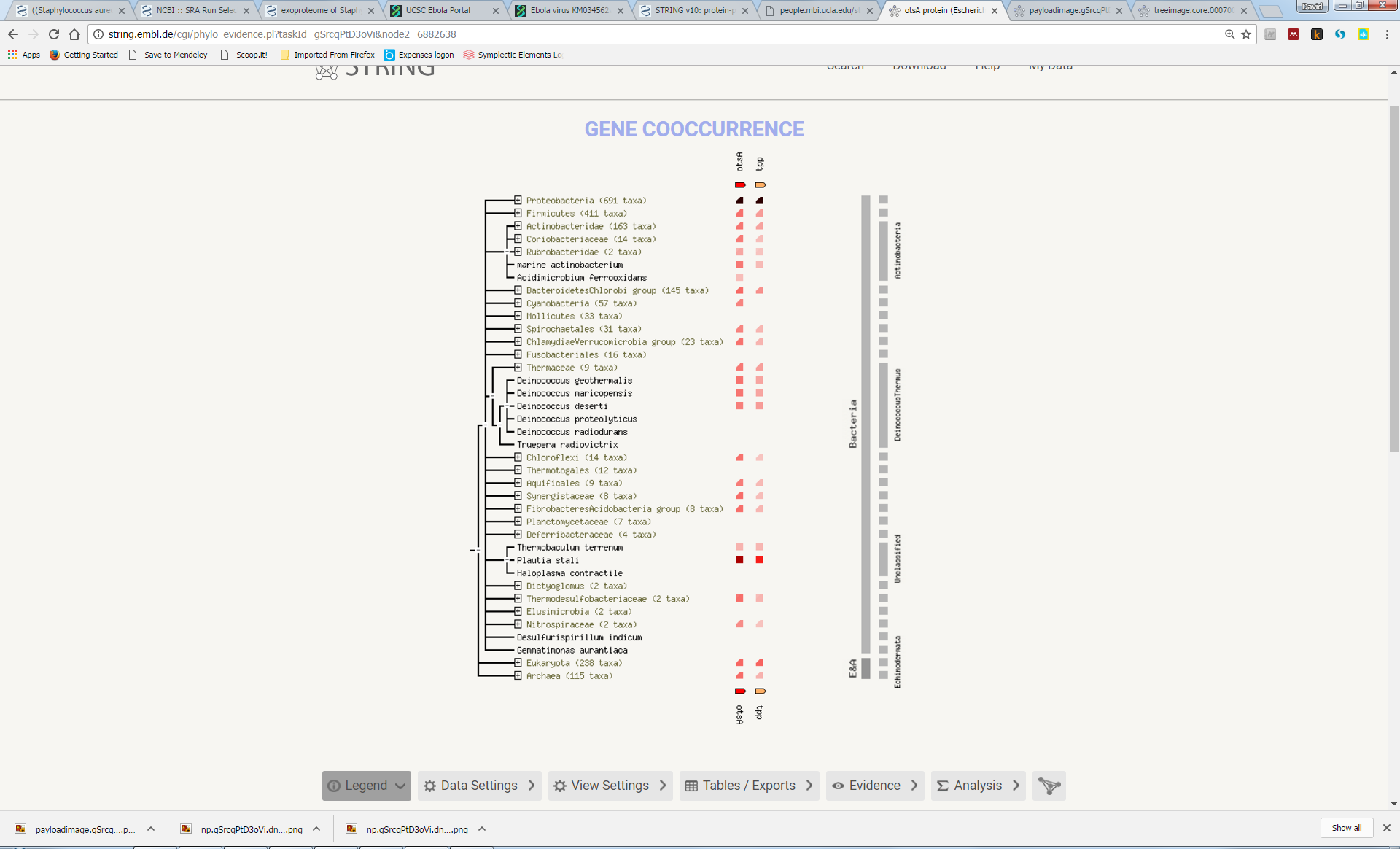
Now, explore some of the other associations, some of which are less obvious, such as the link with katF or osmY.
3.6 Finding and displaying open reading frames (ORFs)
A double-stranded DNA sequence has six different reading frames (three forward and three reverse). An open reading frame (ORF) is the part of a reading frame that has the potential to code for a protein or peptide. An ORF is a continuous stretch of DNA beginning with a start codon usually methionine (ATG) and ending with a stop codon (TAA, TAG or TGA in most genomes).
For this task, you will use the NCBI ORF Finder at http://www.ncbi.nlm.nih.gov/gorf/gorf.html
Exercise: Use this tool to find the ORfs in the human opsin-5 transcript sequence (accession number NM_181744) or in your newly discovered gene.

Exercise: Can you find which ORF encodes the protein product of this transcript? Hint: the predicted amino acid sequence of opsin-5 (accession number NP_859528.1) is:
>gi|38678524|ref|NP\_859528.1| opsin-5 \[Homo sapiens\] MALNHTALPQDERLPHYLRDGDPFASKLSWEADLVAGFYLTIIGILSTFGNGYVLYMSSRRKKKLRPAEIMTINLAVCDLGISVVGKPFTIISCFCHRWVFGWIGCRWYGWAGFFFGCGSLITMTAVSLDRYLKICYLSYGVWLKRKHAYICLAAIWAYASFWTTMPLVGLGDYVPEPFGTSCTLDWWLAQASVGGQVFILNILFFCLLLPTAVIVFSYVKIIAKVKSSSKEVAHFDSRIHSSHVLEMKLTKVAMLICAGFLIAWIPYAVVSVWSAFGRPDSIPIQLSVVPTLLAKSAAMYNPIIYQVIDYKFACCQTGGLKATKKKSLEGFRLHTVTTVRKSSAVLEIHEEWE
So, in your newly discovered gene, can you find a single ORF that encodes the newly discovered protein? If so, then in which reading frame does the ORF fall? If not, then why not? (You may wish to draw a diagram here)
By now, I hope that you are now convinced that it is possible to make new discoveries in biology surprisingly easily by exploring freely available data and using freely available analysis tools. This is thanks to the efforts of software developers, curators and database managers as well as the researchers who deposit their data into these public repositories. The techniques that you explored today can even lead to insights that are worthy of publication in a scientific journal. As an example, here is one of my research papers based entirely on a systematic, yet quite simple, analysis of publicly available DNA and protein sequence data. It didn’t require any expensive equipment or specialised laboratory, just a computer with an internet connection and some curiosity!
:-)